

Abstract
Mice homozygous for a null mutation in the winged helix transcription factor HNF3α showed severe postnatal growth retardation followed by death between P2 and P12. Homozygous mutant mice were hypoglycemic despite unchanged expression of HNF3 target genes involved in hepatic gluconeogenesis. Whereas insulin and corticosteroid levels were altered as expected, plasma glucagon was reduced markedly in the mutant animals despite the hypoglycemia that should be expected to increase glucagon levels. This correlated with a 70% reduction in pancreatic proglucagon gene expression. We also showed that HNF3α could bind to and transactivate the proglucagon gene promoter. These observations invoke a central role for HNF3α in the regulatory control of islet genes essential for glucose homeostasis in vivo.
Keywords: Hepatocyte nuclear factor, endoderm development, metabolic regulation
The HNF3 (hepatocyte nuclear factor 3) proteins were discovered by their ability to bind to the promoters of the genes encoding α-antitrypsin and transthyretin (Costa et al. 1989). Cloning of the cDNAs identified three HNF3 genes (α, β, and γ in mammals; Lai et al. 1990, 1991). Analysis of the crystal structure of HNF3γ showed that the DNA binding, or winged helix domain of the HNF3 proteins has a striking similarity to the linker histones H1 and H5 (Clark et al. 1993). It was shown recently that the HNF3 proteins can reposition nucleosomes in the albumin enhancer (McPherson et al. 1993; Shim et al. 1998). Nucleosome binding by the HNF3 proteins does not compact the chromatin, but is rather correlated with an active albumin enhancer (Cirillo et al. 1998).
All three HNF3 genes are expressed during embryonic development. During formation of the definite endoderm HNF3β is activated first, followed by HNF3α, and finally HNF3γ (Ang et al. 1993; Monaghan et al. 1993; Sasaki and Hogan 1993). In addition, HNF3β is expressed in the node, whereas both HNF3α and HNF3β mRNAs are present in the notochord and floorplate (Ang et al. 1993; Monaghan et al. 1993; Ruiz i Altaba et al. 1993; Sasaki and Hogan 1993). HNF3β is absolutely required for notochord and floorplate formation, because these structures are missing in embryos homozygous for a targeted null mutation in the HNF3β gene (Ang and Rossant 1994; Weinstein et al. 1994). However, because of the early lethality of the homozygous mutant embryos, the role of HNF3β in gut, pancreas and liver development has not yet been assessed. Deletion of the HNF3γ gene leads to a much milder phenotype. HNF3γ−/− mice are viable but show reduced transcription of several HNF3 target genes in the liver (Kaestner et al. 1998).
Recently, the role of HNF3α in metabolism was analyzed in visceral endoderm derived from HNF3α−/− embryoid bodies in vitro (Duncan et al. 1998). In this system, lack of HNF3α led to an increase in the mRNA levels of various serum proteins and glycolytic enzymes, concomitant to an increase in the mRNAs of the transcription factors HNF1α and HNF4α. To test the function of HNF3α in the differentiation of the definite endoderm and in metabolic regulation in vivo, we have generated mice lacking HNF3α via gene targeting. This mutation deletes the entire DNA-binding domain of the HNF3α protein and is therefore considered a null allele. Here we discuss the phenotypic consequences of the mutation on embryonic development as well as hepatic and pancreatic function.
Results
Gene targeting of the HNF3α gene
Mice lacking a functional HNF3α gene product were generated by homologous recombination in mouse embryonic stem (ES) cells. The HNF3α locus had been cloned previously from a mouse strain 129 genomic library to allow for efficient gene targeting (Kaestner et al. 1994a). We constructed a targeting vector that deletes the entire winged helix DNA-binding domain and carboxy-terminal region of the protein and creates an in-frame fusion with the Escherichia coli lacZ gene. Care was taken not to delete any adjacent nontranslated regions, to avoid interfering with the regulation of the HNF3α locus. The complete targeting strategy is depicted in Figure 1A. After electroporation and selection of embryonic stem cells, 290 stably transfected neomycin-resistant ES-cell clones were obtained. We analyzed these clones by probing of Southern blots with a gene fragment not contained within the targeting vector (5′ probe) and found that two clones had undergone homologous recombination (Fig. 1B). Both targeted ES cell clones were shown to be without unintended rearrangements at the HNF3α locus by Southern blot analysis with several probes located 5′, 3′, and internal to the targeting vector (data not shown) and thus contain the HNF3αlacZ allele as schematized in Figure 1A. Germ-line chimeras and mice heterozygous for the HNF3αlacZ mutation were obtained for both ES cell lines. In all parameters studied, both lines gave identical results, indicating that the phenotype observed is indeed caused by the targeted mutation in the HNF3α locus and not caused by another, unrelated mutation derived from the ES-cell clones. In the following, we have combined the results obtained from both lines.
Figure 1.
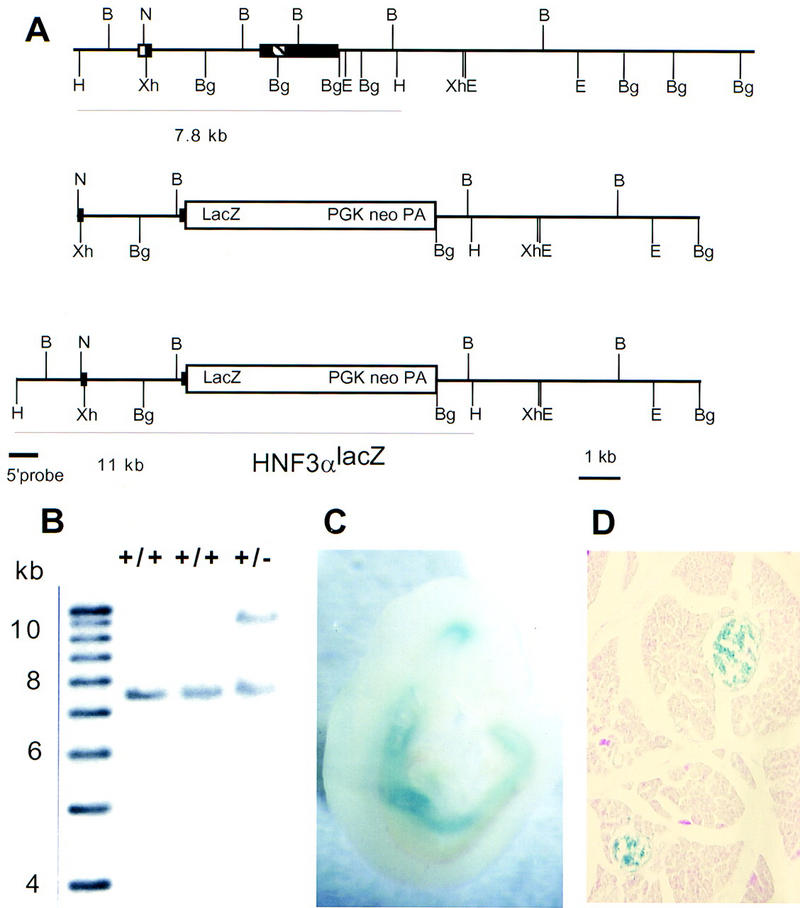
Targeting strategy for HNF3α inactivation. (A) (Top line) Gene structure of the HNF3α locus. Exons are indicated as boxes, the striped box represents the winged helix domain. (Middle line) Targeting vector used for homologous recombination in embryonic stem cells. (Bottom line) Gene structure of the targeted allele (HNF3αlacZ). The 5′ probe was used for the Southern blot shown in B. (B) BamHI; (Bg) BglII; (E) EcoRI; (H) HindIII; (N) NotI; (Xh) XhoI. (B) Southern blot analysis of the correctly targeted ES-cell clone. Genomic ES-cell DNA was digested with HindIII, size-fractionated by agarose gel electrophoresis, blotted onto nylon membrane, hybridized with the 5′ probe, and analyzed by autoradiography. (C) β-Galactosidase staining of a day 9.5 postcoitum embryo heterozygous for the HNF3αlacZ allele indicative of the transcriptional activity of the targeted allele. Expression is observed in the ventral midbrain, the floor plate of the spinal cord, the notochord, foregut, hindgut, and the liver primordium. (D) β-Galactosidase staining of adult pancreas obtained from a mouse heterozygous for the HNF3αlacZ allele. Expression is observed in the entire endocrine pancreas (islet of Langerhans).
We expected that replacement of the HNF3α coding region with the lacZ gene would result in expression of β-galactosidase in all cells that normally express HNF3α. When staining embryos for β-galactosidase activity (Fig. 1C; data not shown), we found expression in the ventral aspects of the midbrain, in the notochord and floor plate of the neural tube, and in the gut tube and liver primordium. These are exactly those tissues that had been shown previously to express HNF3α mRNA by in situ hybridization (Ang et al. 1993; Monaghan et al. 1993; Sasaki and Hogan 1993). Figure 1D demonstrates in addition that in the adult pancreas HNF3α expression is localized to the islets of Langerhans. Comparison of 9.5 postcoitum embryos homozygous for the HNF3αlacZ mutation (HNF3α−/−) to the heterozygous littermates (HNF3α+/-) indicated that despite the early onset of HNF3α expression in the embryo, lack of HNF3α was not detrimental to early embryonic development (data not shown).
Promoter analysis of the HNF3α gene had demonstrated the presence of an HNF3 binding site at position −476/−465 with respect to the start site of transcription in the rat gene (Peterson et al. 1997). This site was shown to bind all three HNF3 proteins (α, β, and γ) in liver nuclear extracts. Deletion of this site resulted in an ∼60% reduction in promoter activity in human hepatoma cells. Therefore, both cross-regulation (by HNF3β and HNF3γ) and autoregulation (by HNF3α itself) were proposed to take part in the regulation of the HNF3α gene. Additional evidence for cross-regulation among the HNF3 genes had come from overexpression of HNF3β in transgenic mice, which led to an induction of HNF3α at the site of ectopic HNF3β expression (Sasaki and Hogan 1993). Furthermore, gene ablation of HNF3γ resulted in elevated mRNA levels for HNF3α and HNF3β in the liver of HNF3γ−/−animals (Kaestner et al. 1998).
Through the design of our targeting vector, we were able to address the issue of autoregulation of the HNF3α gene, as we could assess the transcriptional activity of both the wild-type and the targeted allele. We performed a quantitative RNase protection assay with RNA isolated from livers of postnatal day 8 mice and assessed the mRNA levels for both HNF3α alleles simultaneously. The targeted (HNF3αlacZ) allele was expressed in the homozygous mutants at the same level as the HNF3α allele in wild-type animals, indicating the absence of significant autoregulation for the HNF3α gene in vivo (data not shown). A similar absence of autoregulation at the HNF3α locus had been observed in embryoid bodies deficient for HNF3α (Duncan et al. 1998).
Mice deficient for HNF3α die postnatally
The phenotypic consequences of complete ablation of HNF3α were assessed in the offspring from crosses of heterozygous mice. Fetuses obtained before birth showed no deviation from the expected Mendelian distribution and no apparent morphological abnormalities (data not shown). However, HNF3α is clearly required for postnatal life, as of more than 200 offspring from matings of heterozygous parents, not a single homozygous mutant mouse survived to four weeks of age. Initially, when we analyzed mice in a mixed (129SvEv/C57Bl6) background, we noted a wide variation in the length of survival of the HNF3α−/−animals. The importance of the genetic background on the severity of mutant phenotypes in mice has been well documented (Threadgill et al. 1995). To obtain a more uniform phenotype, we decided to analyze the phenotypic consequences of the HNF3α mutation in several defined genetic backgrounds. To this end, we backcrossed HNF3α heterozygous mice for 10 generations to both 129SvEv and C57BL6 mice (incipient congenic strains, at N10, >99.9% of the genes are homozygous for the alleles represented in the respective parental strain). In addition to the two inbred lines, the effect of the mutation was also assessed in an F1 hybrid between the two lines (129SvEv × C57Bl6). Using F1 hybrids has the advantage of hybrid vigor by complementation of recessive mutations from both parental strains and was recommended by the Banbury Conference on Genetic Background in Mice (1997). At the same time the F1 hybrid is a defined genetic background, as all mice are genetically uniform with the exception of the HNF3α locus. Survival was very short in the two inbred lines (death on P1 or P2), whereas in the F1 hybrid background, homozygous mutant animals survived for 10–14 days. As this background allows for the most thorough analysis of physiological and biochemical parameters, we concentrated our further analyses on the F1 hybrid.
Hypoglycemia in mice deficient for HNF3α is not caused by lack of expression of gluconeogenic enzymes
In litters of newborn mice, we observed a striking growth retardation in animals deficient for HNF3α (Fig. 2A,B). Homozygous mutants are born with the same birth weights as their littermates, but fall behind shortly after birth. Maximal weight is attained between 8 and 10 days. Between 10 and 14 days the HNF3α mutant mice lose weight rapidly and eventually die of severe hypoglycemia (as low as 10 mg/dl) and dehydration. Attempts at prolonging their life through injections or feeding of glucose solution were unsuccessful. This failure to thrive in the homozygous mutant mice is apparently not the result of feeding problems, as the mice have milk in their stomachs at least until day 10 after birth.
Figure 2.
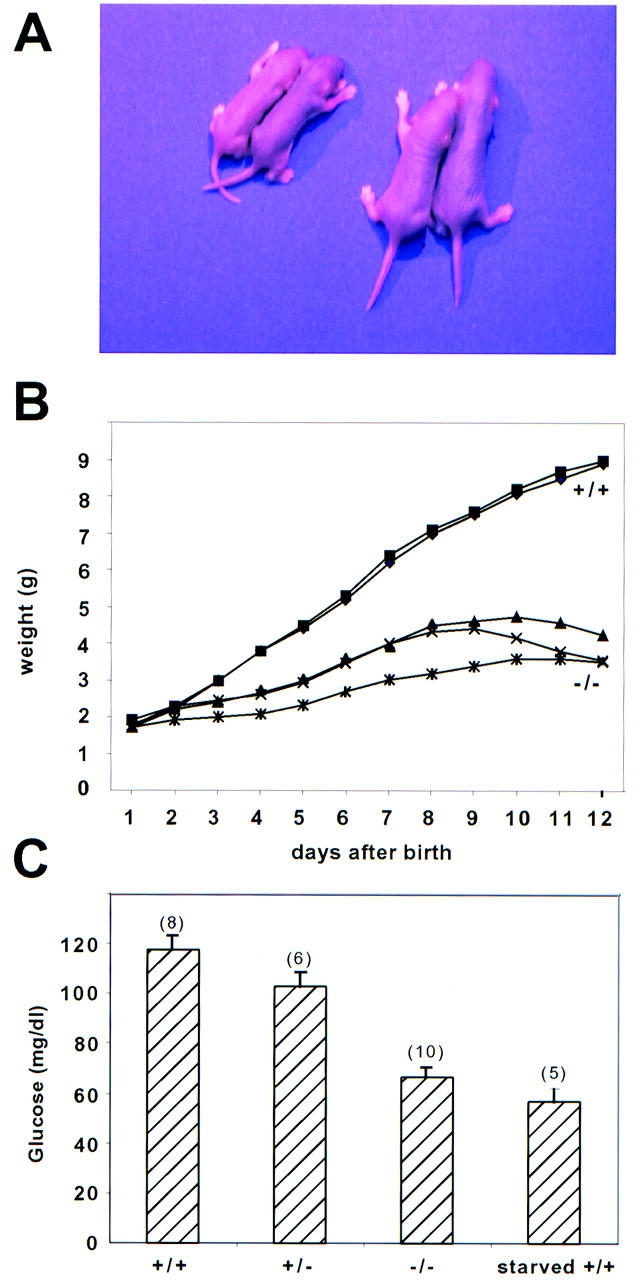
HNF3α−/− mice are growth retarded and hypoglycemic. (A) A typical litter from a cross between HNF3α+/− mice mice (F1 hybrid) on postnatal day 5. The two smaller mice on the left are HNF3α−/−. (B) Growth curves for a representative litter obtained from an intercross between heterozygous mice. The offspring are an F1 hybrid between 129SvEv and C57BL/6. Beyond postnatal day 12 the homozygous mutants lose weight rapidly and die with severe hypoglycemia. (C) Offspring (F1 hybrids) from matings between heterozygous mice were sacrificed on postnatal day 8 and blood glucose was determined. Starved wild-type (+/+) littermates were kept without food for 24 hr before sacrifice. Values are means ± standard error [number (n) indicated in parenthesis], differences between glucose levels were statistically significant (P < 0.001 for +/+ vs. −/−; P < 0.05 for +/+ vs. +/−) by Student’s t-test.
Postnatal death and hypoglycemia were also observed in mice deficient for the transcription factor CCAAT/enhancer-binding protein α (C/EBPα), although in this case the homozygous mutant mice die within the first 8 hr after birth (Wang et al. 1995; Flodby et al. 1996). A hallmark of the C/EBPα null mice is their inability to store glycogen in late gestation, which results in a dramatic fall in blood glucose levels shortly after birth. In contrast, HNF3α−/− mice store glycogen prenatally as evidenced by histological (PAS) staining (data not shown).
As the HNF3 proteins had been shown to be involved in the regulation of several gluconeogenic enzymes in the liver in vitro (for review, see Cereghini 1996), we hypothesized that the growth retardation in the HNF3α mutants might be secondary to hypoglycemia caused by insufficient hepatic gluconeogenesis. We opted to analyze glucose levels in the F1 hybrid mice on postnatal day 8, at a time when the animals are still growing and actively feeding. As is shown in Figure 2C, HNF3α−/− mice are hypoglycemic when compared to their littermates. Interestingly, the glucose levels in the HNF3α−/− mice are very similar to those seen in wild-type littermates that have been starved for 24 hr. Thus, the HNF3α−/− mice behave like starving mice, despite the fact that they have milk in their stomachs.
Hepatic gluconeogenesis is initiated in mammals in the perinatal period in response to dramatic changes in glucagon, corticosteroid, and insulin levels. This leads to a concerted transcriptional activation of several genes encoding gluconeogenic enzymes, several of which also have binding sites for the HNF3 proteins in their promoters or enhancers. These binding sites are relevant for the expression levels of these genes, as both PEPCK and TAT mRNA are reduced in mice lacking the transcription factor HNF3γ (Kaestner et al. 1998). Therefore, we investigated the expression levels of mRNAs encoding gluconeogenic enzymes as well as those of other known HNF3 targets in liver from wild-type and homozygous mutant mice. As is shown in Figure 3A, there are no significant differences in the expression levels for any of the genes analyzed. Thus, the postnatal induction of mRNAs encoding gluconeogenic enzymes in the liver is not impaired in the HNF3α−/− mice and can therefore not explain the hypoglycemia observed in these animals.
Figure 3.

Analysis of steady-state mRNA levels of potential HNF3α targets in liver. (A) Total RNA (10 μg) from livers of wild type (+/+) or homozygous mutants (−/−) was separated on denaturing agarose gels, blotted onto nylon membrane, and hybridized to the probes indicated [(Apo) Apolipoprotein; (SDH) serine dehydrogenase; (PEPCK) phosphoenolpyruvate carboxykinase; (G6Pase) glucose 6 phosphatase; (TAT) tyrosine amino transferase; (TF) transferrin; (TTR) transthyretin; (GLUT2) glucose transporter 2]. Cytochrome c oxidase (COX) served as loading control. The mutant animal in lane 10 was extremely hypoglycemic at the time of sacrifice (<20 mg/dl), and the apparent induction of SDH, TAT, and PEPCK mRNAs is caused by elevated corticosteroid levels. PhosphorImage analysis did not reveal significant differences between the mRNA levels between the wild-type and mutant groups (data not shown). (B) Total RNA (30 μg) from livers of wild type (+/+) or homozygous mutants (−/−) was analyzed by RNase protection assay for mRNA levels of hepatic transcription factors. (HNF) hepatocyte nuclear factor; (TBP) TATA box binding protein. TBP served as loading control.
In several knockouts of transcription factors belonging to gene families, targeted mutation of one gene led to an up-regulation of other transcription factors, demonstrating the existence of regulatory networks between these transcription factors (Rudnicki et al. 1992; Kaestner et al. 1998). In addition, deletion of HNF3α in visceral endoderm differentiated from embryoid bodies in vitro was shown to lead to an increase in the mRNA levels of HNF1α and HNF4α (Duncan et al. 1998). To address the possibility that changes in hepatic transcription factors could play a role in the liver phenotype of the HNF3α−/− mice, we analyzed their steady-state mRNA levels by quantitative RNase protection assay. Consistent with the findings by Duncan et al. (1998), mRNA levels of HNF3β and HNF3γ were unchanged in the livers from mutant animals (data not shown). However, in contrast to the situation in embryoid bodies, there is no change in the transcript levels of HNF1α and HNF4α in liver. Similarly, the relative mRNA levels for HNF1β and HNF6 are unaffected (Fig. 3B).
Our findings regarding the unchanged expression of several hepatic HNF3 targets are at odds with those obtained by Duncan and colleagues in embryoid bodies differentiated from ES cells lacking HNF3α (Duncan et al. 1998). These investigators had observed three- to eightfold up-regulation of steady-state mRNA levels for the apolipoproteins AI, AII, AIV, B, and CII, albumin and the glycolytic enzymes aldolase B and L-pyruvate kinase. In addition, the mRNAs for the transcription factors HNF1α and HNF4α were induced as well. We considered the possibility that embryoid bodies might represent fetal liver or yolk sac more closely than newborn liver. Therefore we analyzed the mRNA levels of selected HNF3 targets in fetal (day 12.5 p.c.) livers and yolk sac by reverse transcription/PCR. As is shown in Figure 4, there was no change in the mRNA abundance of ApoAII, ApoCII, L-pyruvate kinase, and HNF1α in fetal liver or yolk sac from HNF3α mutant mice.
Figure 4.

Analysis of steady-state mRNA levels of potential HNF3α targets in fetal liver and yolk sac. Total RNA from fetal livers or yolk sac of wild type (+/+) or homozygous mutants (−/−) from day 12.5 postcoitum mouse embryos was reverse transcribed and cDNAs amplified by PCR in the presence of [32P]αdATP with primers specific for the genes indicated. After separation on native polyacrylamide gels, the bands were visualized and quantitated using a PhosphoImager. (HPRT) Hypoxanthine-phosphoribosyltransferase; (Apo) apolipoprotein; (HNF) hepatocyte nuclear factor; (L-PK) liver pyruvate kinase.
Hormonal imbalance in mice deficient for HNF3α
An imbalance in the hormonal regulation of glucose homeostasis could also contribute to the hypoglycemia in the HNF3α−/− animals. For instance, hyperinsulinemia could contribute to the hypoglycemia observed. Therefore we analyzed insulin, corticosteroid, and glucagon levels in plasma obtained from postnatal day 8 mice (F1 hybrid background). As is shown in Figure 5A, insulin levels are very low in the HNF3α−/− mice, consistent with their low glucose levels. Conversely, corticosteroid levels are very high in the homozygous mutant mice, which is the appropriate response and an effort of these animals to mobilize gluconeogenesis (Fig. 5B). However, glucagon levels are inappropriately low (Fig. 5C) in the face of hypoglycemia. The paradoxical reduction in circulating glucagon detected in the HNF3α−/− mice is further evident when compared to the higher levels of circulating glucagon in HNF3α+/− and HNF3α+/+ littermate controls, despite the fact that these mice have higher glucose levels, which should be associated normally with a comparatively greater suppression of circulating glucagon in vivo.
Figure 5.
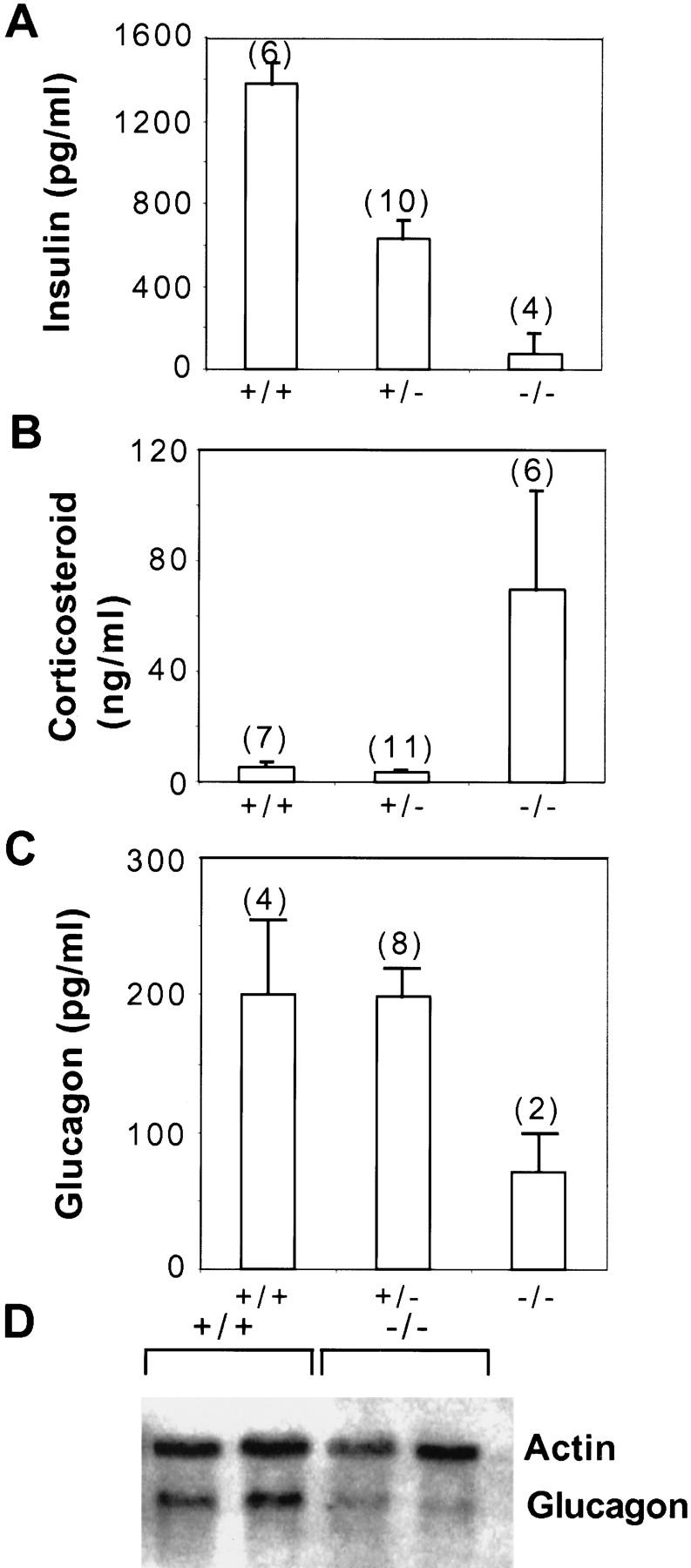
Analysis of hormone levels in HNF3α mutant mice. Offspring (F1 hybrids) from matings between heterozygous mice were sacrificed on postnatal day 8 and plasma hormone levels determined as described in Materials and Methods. The glucagon values for the homozygous mutant mice (−/−) were obtained from plasma pooled from three animals each because of the small size of the mutants. (A) Insulin, (B) corticosteroids, (C) glucagon. Values are means ± standard error (number of n indicated in parenthesis), differences among +/+ or +/− and −/− hormone levels were statistically significant (P < 0.01 for insulin and cortocosteroids; P < 0.05 for glucagon) by Student’s t-test. (D) Glucagon mRNA is reduced in the pancreas of HNF3α mutant mice. Total RNA (10 μg) from pancreas of wild type (+/+) or homozygous mutants (−/−) was separated on denaturing agarose gels, blotted onto nylon membrane and hybridized to the probes indicated.
Several mechanisms could account for this relative glucagon insufficiency in the HNF3α−/− mice, including abnormalities in glucagon synthesis and/or secretion. As HNF3α is a transcription factor, and as an HNF3-binding site had been shown previously to be important in the regulation of glucagon gene expression in tissue culture cells (Philippe et al. 1994), we investigated glucagon mRNA levels in the pancreas of HNF3α−/− mice. As is shown in Figure 5D, glucagon mRNA levels are reduced by ∼70% in the HNF3α mutants. The decrease in pancreatic glucagon mRNA in HNF3α−/− mice with hypoglycemia suggests that a primary defect at the level of glucagon gene expression contributes to the low levels of circulating plasma glucagon and defective counter-regulation to hypoglycemia.
Pancreatic cell lineage allocation in HNF3α−/− mice
The observed decrease in glucagon mRNA expression in the HNF3α−/− mice could be caused either by a change in the glucagon steady-state mRNA levels per glucagon-producing cell or a change in the cell lineage allocation. Lack of HNF3α could, for instance, result in an increase in insulin-producing β cells at the expense of the glucagon-producing α cells. This possibility is relevant in light of recent gene-targeting experiments of other transcription factors expressed in the pancreas (for review, see Habener and Stoffers 1998). For instance, it was shown that Pax6 is required for the differentiation of α cells (St-Onge et al. 1997), whereas mice lacking Pax4 have no β cells (Sosa-Pineda et al. 1997).
Cell lineage allocation was assessed by indirect immunofluorescence staining for α, β, and δ cells using antibodies specific for glucagon, insulin, and somatostatin, respectively. As is shown in Figure 6, cell lineage allocation in the endocrine pancreas of postnatal day 8 HNF3α mutant mice is indistinguishable from that of wild-type littermate controls. Therefore, we conclude that the decrease of glucagon mRNA expression in the HNF3α mutant mice is caused by a change in the relative cellular content of steady-state glucagon mRNA, which in turn is most likely caused by reduced transcriptional activity of the glucagon gene. These observations strongly suggest that HNF3α is an essential regulator of glucagon gene transcription in the endocrine pancreas.
Figure 6.
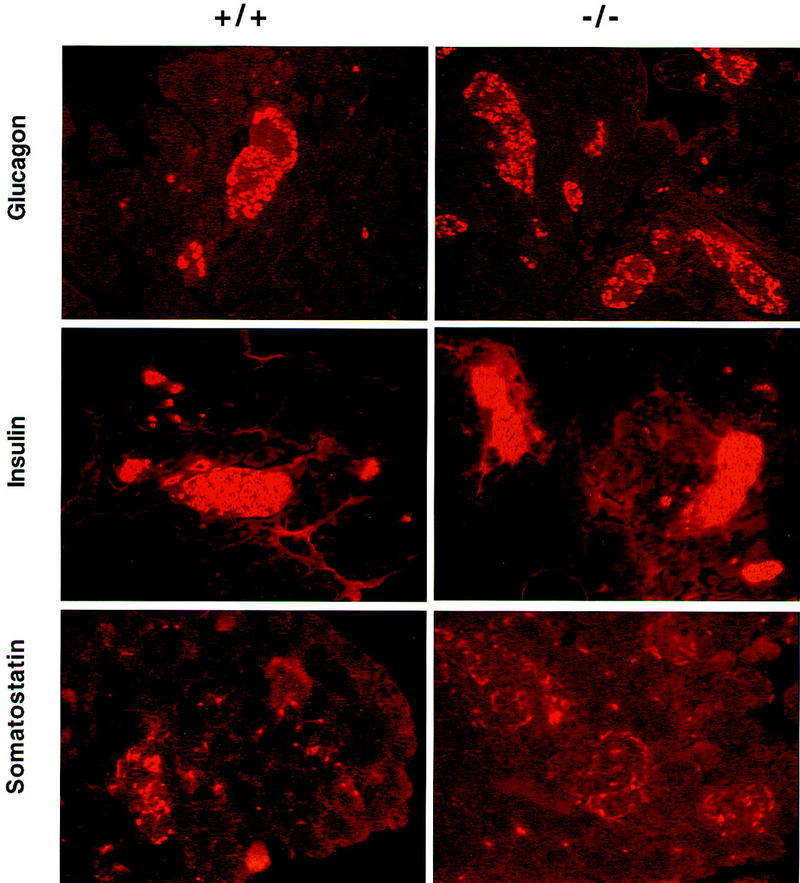
Cell lineage allocation in the pancreas of HNF3α mutant mice. Offspring (F1 hybrids) from matings between heterozygous mice were sacrificed on postnatal day 8 and processed for immunohistochemistry described in Materials and Methods with antisera to the hormones indicated. Tissues are from wild type (+/+) or homozygous mutants (−/−) littermates (postnatal day 8) as indicated.
HNF3α binds to and transactivates the glucagon promoter
HNF3 α, β, and γ transcripts have been detected in the pancreas (Monaghan et al. 1993; Cockell et al. 1995; Vaisse et al. 1997), and more specifically, in glucagon-producing islet cells (Philippe et al. 1994). Our β-galactosidase staining of adult pancreas from HNF3α+/− animals demonstrates expression of HNF3α in the entire pancreatic islet (Fig. 1D). The results of previous studies using cell lines in vitro have suggested a role for the HNF3 proteins in the control of glucagon gene transcription (Philippe et al. 1994). Although HNF3 has been shown to function generally as an activator of gene transcription (Cereghini 1996), HNF3β repressed the activity of glucagon promoter constructs in a transfected islet cell line. Whether this observation might be attributable to nonphysiological overexpression of HNF3β and squelching of promoter activity is not clear, as mice with a targeted deletion in the HNF3β gene die around day E10–E11 (Ang and Rossant 1994; Weinstein et al. 1994), precluding assessment of glucagon gene expression in the endocrine pancreas in the absence of HNF3β.
In contrast to the postulated negative role of HNF3β acting through the glucagon gene G2 element to repress transcription, deletion of the G2-binding site leads to a major loss of glucagon gene transcriptional activity (Philippe et al. 1994), implying that one or more positive factors clearly activate glucagon gene transcription through the G2 enhancer. Furthermore, HNF3β has been shown to activate the proglucagon promoter in transfected HepG2 cells, and protein kinase C-dependent activation of the glucagon promoter appears to be mediated through the positive actions of HNF3β on the glucagon gene G2 element (Furstenau et al. 1997). The finding of reduced glucagon mRNA transcripts in HNF3α mutant mice implicates a previously unrecognized role for HNF3α in the regulation of glucagon gene transcription. To determine whether HNF3α is present in nuclear extracts of glucagon-producing cells and capable of binding the glucagon gene promoter G2 element, we studied HNF3 binding by EMSA using extracts from glucagon-producing cell lines. A major complex was detected with the G2 probe that was reduced and supershifted following incubation with antiserum to HNF3α (data not shown). These observations, together with previous studies of HNF3β in islet A cells, are consistent with previous reports documenting expression of all three HNF3 genes in the pancreas (Monaghan et al. 1993; Cockell et al. 1995; Vaisse et al. 1997).
Next we addressed the question of whether HNF3α can act as transcriptional activator in cotransfection assays with glucagon promoter constructs. Expression vectors encoding either HNF3 α, β, or γ were transfected into BHK fibroblasts together with various glucagon promoter/luciferase reporter constructs. Figure 7 illustrates the results of a representative transfection experiment. All three HNF3 proteins are able to transactivate the −220 glucagon promoter 20- to 40-fold. In contrast, no significant transcriptional activity was observed with the −220 GLU–LUC reporter alone, or when cotransfected with the expression vector pHD. A trimeric G2 site in front of a minimal glucagon promoter (3G2–82GLU–Luc) is activated even more strongly, up to 700-fold by HNF3α. Of the three HNF3 proteins, HNF3α is the most efficient transactivator via the G2 element. In contrast, the identical reporter plasmid containing a mutation in G2 that disrupts HNF3 binding (Fig. 7; Philippe et al. 1994) is transcriptionally inactive in the presence of the cotransfected HNF3 expression plasmids, providing further evidence that transactivation is dependent on the presence of an intact wild-type HNF3-binding site.
Figure 7.

HNF3α transactivates glucagon reporter constructs. Transcriptional activity of rat glucagon promoter–luciferase plasmids in BHK fibroblasts. Reporter plasmids contained the G2 element in its native context within the proximal promoter region of the glucagon gene proximal promoter (−220GLU–LUC), or three copies of the wild-type or mutant G2 element fused to a minimal truncated glucagon promoter (3G2–82GLU–LUC and 3G2M4–82GLU–LUC, respectively). The reporter plasmids were transfected in quadruplicate, alone, or in combination with the expression vector alone, pHD, or with expression vectors encoding HNF3 α, β, or γ. The results obtained represent the results of three separate experiments, are expressed as relative luciferase activity, and are normalized to the luciferase activity obtained following transfection of the respective reporter plasmids alone.
Discussion
We have shown that HNF3α−/− mice obtained by gene targeting die in the postnatal period with hypoglycemia, weakness, and dehydration. This is accompanied by a paradoxical decrease in circulating glucagon levels that is most likely caused by the inappropriately reduced levels of glucagon mRNA in pancreatic α cells. We have thus identified HNF3α as an essential activator of the glucagon gene in vivo. Consistent with this finding, HNF3α binds to the G2 site of the glucagon promoter and activates G2-dependent glucagon promoter activity in vitro. The importance of the HNF3 family for regulation of both islet hormone genes and genes involved hepatic glucose production, taken together with the failure of glucose counterregulation observed in HNF3α−/− mice, strongly suggests an essential role for HNF3α in the transcriptional control of genes essential for glucose homeostasis in vivo.
The results of our EMSA and transfection analyses are consistent with a direct effect of HNF3α on glucagon gene transcription in vitro, and these observations are extended and further validated by the finding of reduced levels of glucagon gene expression in HNF3α−/− mice in vivo. As the glucagon promoter is also regulated in the islets of Langerhans by a diverse number of transcription factors, including Cdx-2/3, Brn4, and Pax6 (Jin and Drucker 1996; Hussain et al. 1997; Sander et al. 1997) an indirect effect of HNF3α on the expression, binding, or activity of these genes/proteins cannot be excluded. Nevertheless, we did not detect evidence for a higher molecular weight HNF3 complex in EMSA studies, and little information is available concerning the regulation of the Pax6, Cdx-2/3, or Brn4 genes in the islet A cell. Furthermore, as HNF3 proteins bind to the G2 promoter element, whereas Pax6, Cdx-2/3, and Brn4 act through the G3 and G1 elements, the available data suggests strongly that HNF3α alone is capable of directly activating glucagon gene transcription in the endocrine pancreas in vivo.
Our data provide strong evidence for the positive action of HNF3 proteins on glucagon gene transcription in that loss of HNF3α is associated clearly with a reduction in glucagon gene expression, even in the face of hypoglycemia. As hypoglycemia has been shown to increase islet glucagon gene expression in vivo (Chen et al. 1989), the paradoxical reduction in levels of pancreatic glucagon mRNA in HNF3α mutant mice despite the presence of hypoglycemia further underscores the essential role of HNF3α as a positive activator of proglucagon gene transcription.
The expression of all three HNF3 genes in the islet A cell raises the possibility that these proteins compete with each other for binding to specific target sites in HNF3-responsive promoters. Recent evidence from analysis of embryoid bodies with inactivated HNF3α or HNF3β alleles provides further support for this hypothesis by demonstrating that the ratio of HNF3α or HNF3β is an important determinant of the relative levels of gene transcription (Duncan et al. 1998). Furthermore, the ratio of HNF3α to HNF3β is regulated by insulin in embryoid bodies, providing further evidence for a link between islet function and the expression of HNF3 target genes.
There is an obvious discrepancy between our finding regarding the expression of HNF3 targets in yolk sac and fetal and newborn liver devoid of HNF3α and those obtained by Duncan et al. (1998) in visceral endoderm differentiated from embryoid bodies deficient for HNF3α. Whereas there was no change in steady-state mRNA levels for any of the HNF3 targets analyzed in liver and yolk sac, some of the same genes were found to be up-regulated three- to eightfold in visceral endoderm in culture. Thus, it appears that visceral endoderm differentiated from embryoid bodies in vitro does not reflect entirely the situation in the developing liver and yolk sac.
Materials and methods
Gene targeting
λ phage clones containing the murine HNF3α gene had been isolated from a mouse embryonic stem cell (strain 129) library previously (Kaestner et al. 1994a). A gene-targeting vector was constructed in β-galactosidase-containing plasmid pHM2 (Kaestner et al. 1994b). A 2-kb fragment of HNF3α gene was replaced by the lacZ and neomycin-resistance cassette of pHM2. Thereby an in-frame fusion was created between the 89 amino-terminal amino acids of the HNF3α protein and the β-galactosidase protein. The targeting vector deletes most of exon 2 including the entire DNA-binding domain of HNF3α. The targeting vector was linearized with NotI and 20 μg of DNA electroporated into 107 E14-1 embryonic stem cells (Kühn et al. 1991). Stably transfected cells were isolated after selection in 350 μg/ml G418 (GIBCO) and 290 clones analyzed by Southern blot for homologous recombination. A 0.8-kb HindIII/BamHI fragment (5′ probe in Fig. 1) located 5′ to the gene was used as an external probe for Southern analysis of DNA digested with HindIII. Positively targeted clones were confirmed with a probe fragment encoding the neomycin phosphotransferase gene and with three additional restriction digests (data not shown). ES cells from the two correctly targeted clones were injected into blastocysts derived from C57BL/6 mice. Blastocysts were transferred to pseudopregnant NMRI females and chimeric offspring were identified by the presence of agouti hair. Chimeric males were mated to C57BL/6 females to obtain ES-derived offspring that were analyzed by Southern blot of tail DNA to identify the heterozygous (HNF3α+/−) mutants. Germ-line chimeras were backcrossed to both 129SvEv and C57BL/6 mice for 10 generations (N10). Heterozygotes were mated inter se to generate mutant (−/−) mice. Embryos and mice were also genotyped by PCR using three primers: HNF3α 5′ (CTCCGGCCTGGGCTCTATGAAC), HNF3α 3′ (GCCCATGGAGCCCATGCCTCC) and lacZ (CGCCATTCGCCATTCAGGCTGC). PCR reactions were carried out for 32 cycles (94°C, 45 sec; 67°C, 45 sec; 72°C, 90 sec) in a buffer containing 1.5 mm MgCl2.
β-Galactosidase staining
Embryos (day 9.5 p.c.) or adult tissues were dissected in ice-cold PBS and the yolk sac saved for DNA preparation and genotyping by PCR. The embryos were fixed in 4% formaldehyde for 30 min at 4°C, whereas the adult tissues were fixed for 4 hr. Subsequently, embryos and tissues were washed twice in PBS and then incubated in staining solution (4 mm K3[Fe(CN)6]; 4 mm K4[Fe(CN)6]; 0.02% NP-40; 0.01% Na-deoxycholate; 5 mm EGTA; 2 mm MgCl2, and 0.4 mg/ml 5-bromo-4-chloro-3-indolyl-d-galactopyranoside). Prior to sectioning, tissues were dehydrated through an ethanol series, cleared in isopropanol, and embedded in paraffin.
RNA analysis
Total RNA was isolated after homogenization in guanidinium thiocyanate (Chomczynski and Sacchi 1987). RNA was separated in formaldehyde-containing agarose gels for Northern analysis as described previously (Kaestner et al. 1998). Hybond N filters (Amersham) were hybridized in 50% formamide, 5× SSC, 50 mm Na phosphate at pH 6.5, 8× Denhardt’s solution, 1% SDS, and 0.5 mg/ml of total yeast RNA with the probes indicated. RNase protection analysis was carried using the RPAII kit (Ambion). The RNA fragments obtained were separated on denaturing 6% acrylamide gels and the radioactive bands visualized using a PhosphoImager (Molecular Dynamics). RT–PCR analysis was performed as described in Duncan et al. (1998).
Glucose and hormone assays
Newborn mice (postnatal day 8) were sacrificed by decapitation and blood instantly mixed with 5% (vol/vol) of TEL solution (trasylol 10,000 KIU/ml, 60 mm EDTA, 0.5 mg/ml leupeptin). After centrifugation, the resulting plasma was stored at −80°C until assayed. Glucose was determined using glucose test strips (Glucometer Elite, Bayer). Insulin was assayed using ELISA (Crystal Chem), whereas corticosteroids and glucagon were determined using radioimmunassay (ICN Pharmaceuticals and University of Pennsylvania Diabetes Center, respectively).
Immunohistochemistry
Tissues were fixed in 4% paraformaldehyde, embedded in paraffin, and 5-μm sections were applied to Probe-on Plus slides (Fisher Scientific). Deparaffinized and rehydrated slides were permeabilized in 0.2% Triton X-100 for 5 min, then blocked in PBS containing 1% BSA for 30 min at 37°C. The primary antibodies, 1:100 guinea pig anti-insulin (Biogenesis), 1:200 rabbit anti-glucagon (Cambridge Research Biochemicals), or 1:200 rabbit anti-somatostatin (Genosys) were diluted in PBS containing 1% BSA and incubated for 1 hr at 37°C. Slides were washed in PBS containing 1% BSA, then incubated for 45 min at 37°C in the appropriate secondary antibody, rhodamine-conjugated goat anti-guinea pig (ICN) or rhodamine-conjugated goat anti-rabbit (ICN), diluted 1:50 in PBS containing 1% BSA. Slides were washed in PBS, mounted, and viewed by fluorescent microscopy.
Cotransfection assays
LUC reporter constructs −220 GLU–LUC and −82GLU–LUC have been described previously (Jin et al. 1997). 3G2–82GLU–LUC and 3G2M4–82LUC were constructed by annealing synthetic oligonucleotides G2, and G2M4, and three copies of the annealed double-stranded oligonucleotides were ligated, in a 5′–3′ orientation, into the −82GLU–LUC vector. The exact sequences of 3G2–82PLuc and 3G2M4–82GLU–LUC clone were confirmed by DNA sequencing. The mouse HNF3 cDNAs (Kaestner et al. 1994a) were subcloned into the expression vectors pHD (Muller et al. 1988). BHK fibroblasts were grown and transfected as described previously (Jin and Drucker 1996; Jin et al. 1997) using 6 μg of the various luciferase reporter plasmids and 6 μg of either pBluescript (carrier DNA) or 6 μg of the various HNF3 expression plasmids. All transfections were carried out in quadruplicate on at least three separate occasions. Cells were harvested 36 hr later, for analysis of luciferase activity.
Acknowledgments
We thank P.A. Labosky for advice on β-galactosidase staining, S.A. Duncan for supplying protocols and primers for the RT–PCR analysis, and J.A. Blendy and M.A. Lazar for critical reading of the manuscript. We are greatful to S. Ridder, H. Kern, S. Dutton Sackett, and A. Sukman for expert technical assistance. We acknowledge support from the University of Pennsylvania Diabetes Center (P30 DK19525) for performing radioimmunoassays, and the Center for Molecular Studies in Digestive and Liver Disease at the University of Pennsylvania (P30 DK50306). This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases (RO1 DK55342-01 to K.H.K.), the Deutsche Forschungsgemeinschaft through SFB 229 (G.S.), the Medical Research Council of Canada (D.J.D.) and a Canadian Diabetes Association Fellowship (Y.L.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL kaestner@mail.med.upenn.edu; FAX (215) 573-5892.
References
- Ang SL, Rossant J. HNF-3 beta is essential for node and notochord formation in mouse development. Cell. 1994;78:561–574. doi: 10.1016/0092-8674(94)90522-3. [DOI] [PubMed] [Google Scholar]
- Ang SL, Wierda A, Wong D, Stevens KA, Cascio S, Rossant J, Zaret KS. The formation and maintenance of the definitive endoderm lineage in the mouse: Involvement of HNF3/forkhead proteins. Development. 1993;119:1301–1315. doi: 10.1242/dev.119.4.1301. [DOI] [PubMed] [Google Scholar]
- Banbury Conference on Genetic Background in Mice. Mutant mice and neuroscience: Recommendations concerning genetic background. Neuron. 1997;19:755–759. doi: 10.1016/s0896-6273(00)80958-7. [DOI] [PubMed] [Google Scholar]
- Cereghini S. Liver-enriched transcription factors and hepatocyte differentiation. FASEB J. 1996;10:267–282. [PubMed] [Google Scholar]
- Chen PP, Siminovitch KA, Olsen NJ, Erger RA, Carson DA. A highly informative probe for two polymorphic Vh gene regions that contain one or more autoantibody-associated Vh genes. J Clin Invest. 1989;84:706–710. doi: 10.1172/JCI114218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Cirillo LA, McPherson CE, Bossard P, Stevens K, Cherian S, Shim EY, Clark KL, Burley SK, Zaret KS. Binding of the winged-helix transcription factor HNF3 to a linker histone site on the nucleosome. EMBO J. 1998;17:244–254. doi: 10.1093/emboj/17.1.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark KL, Halay ED, Lai E, Burley SK. Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature. 1993;364:412–420. doi: 10.1038/364412a0. [DOI] [PubMed] [Google Scholar]
- Cockell M, Stolarczyk D, Frutiger S, Hughes GJ, Hagenbuchle O, Wellauer PK. Binding sites for hepatocyte nuclear factor 3 beta or 3 gamma and pancreas transcription factor 1 are required for efficient expression of the gene encoding pancreatic alpha-amylase. Mol Cell Biol. 1995;15:1933–1941. doi: 10.1128/mcb.15.4.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa RH, Grayson DR, Darnell JJ. Multiple hepatocyte-enriched nuclear factors function in the regulation of transthyretin and alpha 1-antitrypsin genes. Mol Cell Biol. 1989;9:1415–1425. doi: 10.1128/mcb.9.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan SA, Navas MA, Dufort D, Rossant J, Stoffel M. Regulation of a transcription factor network required for differentiation and metabolism. Science. 1998;281:692–695. doi: 10.1126/science.281.5377.692. [DOI] [PubMed] [Google Scholar]
- Flodby P, Barlow C, Kylefjord H, Ahrlund-Richter L, Xanthopoulos KG. Increased hepatic cell proliferation and lung abnormalities in mice deficient in CCAAT/enhancer binding protein α. J Biol Chem. 1996;271:24753–24760. doi: 10.1074/jbc.271.40.24753. [DOI] [PubMed] [Google Scholar]
- Furstenau U, Schwaninger M, Blume R, Kennerknecht I, Knepel W. Characterization of a novel protein kinase C response element in the glucagon gene. Mol Cell Biol. 1997;17:1805–1816. doi: 10.1128/mcb.17.4.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habener JF, Stoffers DA. A newly discovered role of transcription factors involved in pancreas development and the pathogenesis of diabetes mellitus. Proc Assoc Am Physicians. 1998;110:12–21. [PubMed] [Google Scholar]
- Jin T, Drucker DJ. Activation of proglucagon gene transcription through a novel promoter element by the caudal-related homeodomain protein cdx-2/3. Mol Cell Biol. 1996;16:19–28. doi: 10.1128/mcb.16.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin T, Trinh DK, Wang F, Drucker DJ. Tha caudal homeobox protein cdx-2/3 activates endogenous proglucagon gene expression in InR1-G9 islet cells. Mol Endocrinol. 1997;11:203–209. doi: 10.1210/mend.11.2.9882. [DOI] [PubMed] [Google Scholar]
- Hussain MA, Lee J, Miller CP, Habener JF. POU domain transcription factor brain 4 confers pancreatic a-cell-specific expression of the proglucagon gene through interaction with a novel proximal promoter G1 element. Mol Cell Biol. 1997;17:7186–7194. doi: 10.1128/mcb.17.12.7186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaestner KH, Hiemisch H, Luckow B, Schütz G. The HNF-3 gene family of transcription factors in mice: Gene structure, cDNA sequence, and mRNA distribution. Genomics. 1994a;20:377–385. doi: 10.1006/geno.1994.1191. [DOI] [PubMed] [Google Scholar]
- Kaestner KH, Montoliu L, Kern H, Thulke M, Schütz G. Universal beta-galactosidase cloning vectors for promoter analysis and gene targeting. Gene. 1994b;148:67–70. doi: 10.1016/0378-1119(94)90234-8. [DOI] [PubMed] [Google Scholar]
- Kaestner KH, Hiemisch H, Schütz G. Targeted disruption of the gene encoding hepatocyte nuclear factor 3γ results in reduced transcription of hepatocyte-specific genes. Mol Cell Biol. 1998;18:4245–4251. doi: 10.1128/mcb.18.7.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn R, Rajewski K, Müller W. Generation and analysis of interleukin-4 deficient mice. Science. 1991;254:707–710. doi: 10.1126/science.1948049. [DOI] [PubMed] [Google Scholar]
- Lai E, Prezioso VR, Smith E, Litvin O, Costa RH, Darnell JE., Jr HNF-3A, a hepatocyte-enriched transcription factor of novel structure is regulated transcriptionally. Genes & Dev. 1990;4:1427–1436. doi: 10.1101/gad.4.8.1427. [DOI] [PubMed] [Google Scholar]
- Lai E, Prezioso VR, Tao WF, Chen WS, Darnell JE., Jr Hepatocyte nuclear factor 3 α belongs to a gene family in mammals that is homologous to the Drosophila homeotic gene fork head. Genes & Dev. 1991;5:416–427. doi: 10.1101/gad.5.3.416. [DOI] [PubMed] [Google Scholar]
- McPherson CE, Shim EY, Friedman DS, Zaret KS. An active tissue-specific enhancer and bound transcription factors existing in a precisely positioned nucleosomal array. Cell. 1993;75:387–398. doi: 10.1016/0092-8674(93)80079-t. [DOI] [PubMed] [Google Scholar]
- Monaghan AP, Kaestner KH, Grau E, Schütz G. Postimplantation expression patterns indicate a role for the mouse forkhead/HNF-3 α, β, and γ genes in determination of the definitive endoderm, chordamesoderm and neuroectoderm. Development. 1993;119:567–578. doi: 10.1242/dev.119.3.567. [DOI] [PubMed] [Google Scholar]
- Müller G, Ruppert S, Schmid E, Schütz G. Functional analysis of alternatively spliced tyrosinase gene transcripts. EMBO J. 1988;7:2723–2730. doi: 10.1002/j.1460-2075.1988.tb03126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson RS, Clevidence DE, Ye H, Costa RH. Hepatocyte nuclear factor-3 α promoter regulation involves recognition by cell-specific factors, thyroid transcription factor-1, and autoactivation. Cell Growth Differ. 1997;8:69–82. [PubMed] [Google Scholar]
- Philippe J, Morel C, Prezioso VR. Glucagon gene expression is negatively regulated by hepatocyte nuclear factor 3 β. Mol Cell Biol. 1994;14:3514–3523. doi: 10.1128/mcb.14.5.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnicki MA, Braun T, Hinuma S, Jaenisch R. Inactivation of MyoD in mice leads to up-regulation of the myogenic HLH gene Myf-5 and results in apparently normal muscle development. Cell. 1992;71:383–390. doi: 10.1016/0092-8674(92)90508-a. [DOI] [PubMed] [Google Scholar]
- Ruiz i Altaba A, Prezioso VR, Darnell JE, Jessell TM. Sequential expression of HNF-3 β and HNF-3 α by embryonic organizing centers: The dorsal lip/node, notochord and floor plate. Mech Dev. 1993;44:91–99. doi: 10.1016/0925-4773(93)90060-b. [DOI] [PubMed] [Google Scholar]
- Sander M, Neusbuser A, Kalamaras J, Ee HC, Martin GR, German MS. Genetic analysis reveals that PAX6 is required for normal transcription of pancreatic hormone genes and islet development. Genes & Dev. 1997;11:1662–1673. doi: 10.1101/gad.11.13.1662. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Hogan BL. Differential expression of multiple fork head related genes during gastrulation and axial pattern formation in the mouse embryo. Development. 1993;118:47–59. doi: 10.1242/dev.118.1.47. [DOI] [PubMed] [Google Scholar]
- Shim EY, Woodcock C, Zaret KS. Nucleosome positioning by the winged helix transcription factor HNF3. Genes & Dev. 1998;12:5–10. doi: 10.1101/gad.12.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa-Pineda B, Chowdhury K, Torres M, Oliver G, Gruss P. The Pax4 gene is essentisl for differentiation of insulin-producing β cells in the mammalian pancreas. Nature. 1997;386:399–402. doi: 10.1038/386399a0. [DOI] [PubMed] [Google Scholar]
- St-Onge L, Sosa-Pineda B, Chowdhury K, Mansouri A, Gruss P. Pax6 is required for differentiation of glucagon-producing α cells in mouse pancreas. Nature. 1997;387:406–409. doi: 10.1038/387406a0. [DOI] [PubMed] [Google Scholar]
- Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D, LaMantia C, Mourton T, Herrup K, Harris RC, et al. Targeted disruption of mouse EGF receptor: Effect of genetic background on mutant phenotype. Science. 1995;269:230–234. doi: 10.1126/science.7618084. [DOI] [PubMed] [Google Scholar]
- Vaissa C, Kim J, Espinosa R, III, LeBeau MM, Stoffel M. Pancreatic islet expression studies and polymorphic DNA markers in the genes encoding hepatocytenuclear factor-3α, -3β, -3γ, -4γ, and -6. Diabetes. 1997;46:1364–1367. doi: 10.2337/diab.46.8.1364. [DOI] [PubMed] [Google Scholar]
- Wang ND, Finegold MJ, Bradlet A, Ou CN, Abdelsayed SV, Wilde MD, Taylor LR, Wilson DR, Dralington GJ. Impaired energy homeostasis in C/EBPalpha knockout mice. Science. 1995;269:1108–1112. doi: 10.1126/science.7652557. [DOI] [PubMed] [Google Scholar]
- Weinstein DC, Ruiz i Altaba A, Chen WS, Hoodless P, Prezioso VR, Jessell TM, Darnell JE., Jr The winged-helix transcription factor HNF-3 β is required for notochord development in the mouse embryo. Cell. 1994;78:575–588. doi: 10.1016/0092-8674(94)90523-1. [DOI] [PubMed] [Google Scholar]