

SUMMARY
Neurotrophic small molecules have the potential to aid in the treatment of neuronal injury and neurodegenerative diseases. The natural product fellutamide B, originally isolated from Penicillium fellutanum, potently induces nerve growth factor (NGF) release from fibroblasts and glial-derived cells, although the mechanism for this neurotrophic activity has not been elucidated. Here, we report that fellutamide B potently inhibits proteasome catalytic activity. High resolution structural information obtained from co-crystallization of the 20S proteasome reveals novel aspects regarding β-subunit binding and adduct formation by fellutamide B to inhibit their hydrolytic activity. We demonstrate that fellutamide B and other proteasome inhibitors increased NGF gene transcription via a cis-acting element (or elements) in the promoter. These results demonstrate an unrecognized connection between proteasome inhibition and NGF production, suggesting a possible new strategy in the development of neurotrophic agents.
The development of neurotrophic therapeutics for treatment of neuronal injury or the neurodegenerative effects of stroke, ischemia and CNS diseases (e.g. Parkinson’s Disease, Alzheimer’s Disease) has attracted much attention. In particular, the neuroprotective and restorative effects of nerve growth factor or other neurotrophins (reviewed in (Huang and Reichardt, 2003)) in ameliorating the symptoms or pathophysiology in animal models of the disease(s) have been documented in the literature (Castellanos-Ortega et al., 1999). For example, co-transplantation of NGF along with fetal ventral mesencephalic cells into the striatum of lesioned rats (a model of Parkinson's Disease) significantly restored spontaneous locomotor activity and striatal and nigral dopamine levels compared to those in rats receiving transplanted cells alone (Chaturvedi et al., 2006). These results suggested that NGF exhibited neuroprotective effects on the transplanted cells as well as helped rescue remaining host dopaminergic neurons from cell death. Similarly, NGF can attenuate lesion-induced cholinergic deficits and cognitive impairments in animal models of Alzheimer’s Disease: chronic NGF treatment induced increased blood flow and nicotine uptake in the cerebral cortex (Lapchak, 1993), and implantation of genetically engineered NGF-secreting fibroblasts into early Alzheimer’s patients brains significantly slowed cholinergic nerve deterioration (Tuszynski et al., 2005). However, the requirement for direct administration of neurotrophins and/or large molecules into the CNS in order to circumvent the blood-brain barrier severely limits their therapeutic utility, and is not without side effects (Venero et al., 1996). Efforts to promote penetration of NGF across the blood-brain barrier by conjugating it to transferrin (Liao et al., 2001) have been made; however, the amount of modified NGF detected in the CNS was very small.
The development of small molecule neurotrophic compounds capable of entering the brain is, therefore, an attractive therapeutic strategy. Literature reports have described small molecules that are able to upregulate selected neuronal proteins and/or induce neurite outgrowth of cultured preneuronal cells (Cheng et al., 2006; Warashina et al., 2006). However, the mechanism(s) whereby such molecules act are yet unknown, nor is the extent to which they can completely mimic the actions of endogenous neurotrophins. The fellutamides are marine fungal metabolites (Shigemori et al., 1991) reported to induce the synthesis and secretion of NGF (Yamaguchi et al., 1993) from cultured brain cells and fibroblasts. Thus, fellutamide B might be considered an “indirect neurotrophin”, in much the same manner as a drug like reserpine, which triggers the release of norepinephrine from presynaptic vesicles, is considered an indirect adrenergic agonist. An indirect neurotrophic small molecule is likely to reap the combined benefits of easier CNS access and as well the ultimate therapeutic effects of the induced endogenous proteins. We recently reported on the first total synthesis of fellutamide B (Schneekloth et al., 2006). Here, we explore the mechanism by which fellutamide B exerts its neurotrophin-inducing effect. We show that fellutamide B potently inhibits the 20S proteasome leading to increased NGF gene expression and secretion.
RESULTS
Functional and structural evidence of proteasome inhibition by fellutamide B
Given the similarity in chemical structure between fellutamide B and peptide aldehyde proteasome inhibitors (as exemplified by MG132, see Figure 1a), we investigated whether fellutamide B could inhibit the three hydrolytic activities of the 20S proteasome. As shown in Figure 1b, fellutamide B potently inhibits the chymotryptic-like activity with an IC50 value of 9.4 ± 2.5 nM. The tryptic-like and caspase-like activities were also inhibited by fellutamide B, albeit less potently (2.0 ± 0.4 µM and 1.2 ± 0.8 µM , respectively). The potency of fellutamide B against the chymotryptic-like activity was, in fact, greater than that of the peptide aldehyde inhibitor MG132 (40 ± 3.3 nM) and approached the high potency seen with the irreversible proteasome inhibitor (Meng et al., 1999b) epoxomicin (5.7 ± 1.3 nM) (Figure 1c). As with the other two established proteasome inhibitors, treatment of L-M mouse fibroblasts with fellutamide B led to accumulation of ubiquitinated proteins (Figure 1d), confirming its ability to inhibit the proteasome in intact cells.
Figure 1. Inhibition of proteasome activities by fellutamide B and other inhibitors.



A. Chemical structures of fellutamide B and the known peptide-aldehyde proteasome inhibitor, MG132. Their respective active aldehyde groups are boxed in red. B. Fellutamide B inhibits the chymotryptic-like, the tryptic-like and the caspase-like activities of the mammalian proteasome. Proteolytic reactions were initiated by addition of proteasomes to pre-mixed substrate and inhibitor. Results presented are the mean ± standard error of three independent experiments. C. Potency of fellutamide B versus other proteasome inhibitors to inhibit chymotryptic-like activity of mammalian proteasome. Proteolytic reactions were initiated by addition of proteasomes to pre-mixed substrate and inhibitor. Results presented are the mean ± standard error of three independent experiments. D. Fellutamide B treatment causes accumulation of ubiquitinated proteins in vivo similar to other proteasome inhibitors (top panel). L-M cells were treated for 2 hr. with either 10 µM fellutamide B, 500 nM epoxomicin, 25 µM MG 132 or 0.1% DMSO (vehicle control); corresponding α-tubulin levels (bottom panel).
We next determined the crystal structure of the yeast 20S proteasome in complex with fellutamide B to reveal its mechanism of inhibition. The electron density was well defined in all active sites, showing fellutamide B covalently bound to each active site by a hemiacetal bond formed between its functional aldehyde group and the Thr1 γ-oxygen (Figure 2a), similar to other aldehyde inhibitors, such as calpain inhibitor I, for which the crystal structure is also well-defined (Groll et al., 1997; Löwe et al., 1995). However, the carbonyl-oxygen of the hemiacetal formed by fellutamide B is hydrogen-bridged to the Thr1 N-terminus (2.9 Å); this differs from the interaction between the carbonyl oxygen of calpain inhibitor I and the backbone amide of Gly47 (Figure 2b). This difference is especially noteworthy since previous data on aldehyde inhibitors had defined the backbone amide of Gly47 as the stereotypical oxyanion hole stabilizing the transition state intermediate by hydrogen bonding to the carbonyl group of the peptide bond undergoing hydrolysis.
Figure 2. Structural data of fellutamide B co-crystallized with S. cerevisiae 20S proteasome.


A. Fellutamide B (yellow) bound to the chymotryptic-like subunit (β5) of the proteasome (space filling model). B. Distinct stabilization of hemiacetal carbonyl-oxygen (red) of fellutamide B (green) by Thr1 versus that of calpain inhibitor I (yellow) by Gly47. The chymotryptic-like subunit is grey and the hemiacetal bond itself is colored pink. The inhibitor hydrogen-bonding elements within the chymotryptic-like subunit -- Thr1 and Gly47 -- are colored black. C. Different, subunit-specific orientations of the aliphatic tail of fellutamide B. Fellutamide B is colored green, yellow and blue, when bound to the chymotryptic-like (β5), tryptic-like (β1) and caspase-like (β2) proteasomal subunits, respectively. D. Electron density diagram showing fellutamide B (green) interacting with designated residues (black) along the specificity pocket of the chymotryptic-like active site of the proteasome.
It is known that aldehydes act as proteasome inhibitors only if they contain a peptide backbone that allows their stabilization at the proteasomal active sites through formation of an anti-parallel β-sheet structure. These interactions are necessary to increase the mean residence time of the ligand at the active center. By varying their peptide backbone, proteasome inhibitors sharing the same reactive functional group show preference for certain active sites (Myung et al., 2001). Compared to other aldehyde proteasome inhibitors, fellutamide B also contains an extended β-hydroxy aliphatic tail. Surprisingly, the whole aliphatic tail, which shows high flexibility in solution, has well defined electron density at all proteolytic active sites. This stands in contrast to synthetic long chain aldehyde inhibitors, which are structurally ordered only in their first three residues (Loidl et al., 1999). Interestingly, structural superposition of fellutamide B bound to the proteolytic active sites indicates that while the peptide backbone adopts similar conformations, the orientation of the aliphatic tail differs completely (Figure 2c). In particular, and uniquely at the chymotryptic-like subunit, C24 to C29 of the fellutamide B aliphatic tail (see Figure 1a) interact with a hydrophobic groove through van der Waals forces with protein residues Pro95, Tyr96, Pro115 and Val116 of subunit β6 (Figure 2d). Interestingly, the hydrophobic groove to which the aliphatic tail is bound is formed only during ligand binding, following concerted movements of the aliphatic tail and protein side chain residues.
Neurotrophic effects of fellutamide B and other proteasome inhibitors on mammalian cells
Fellutamide B induces NGF secretion from a number of cells that previously have been used as models to study NGF synthesis and secretion (Table 1). All five of these cell lines (L-M, NIH-3T3, S-180, A172 and C6-2B cells) responded to 10 µM fellutamide B treatment by upregulating NGF secretion, as detected by ELISA of culture medium. L-M cells secreted the most NGF overall and also exhibited the most robust response to the drug, a 16.8-fold average induction, while NIH-3T3 cells also responded to fellutamide B with an average 15.8-fold increase in NGF secretion. S-180 sarcoma cells increased NGF secretion by only 4.0-fold, although perhaps these tumor cells by their very nature are already operating at near their maximum capacity to produce this neurotrophin. By comparison, the two glial-derived cell lines, A172 and C6-2B, secreted NGF on a much smaller scale; however, both responded to fellutamide B by increasing their NGF production 4.9-fold and 6.6-fold, respectively. Not every cell type tested responded to fellutamide B. KB epidermal carcinoma cells and P-388 leukemia cells were used in the original report on the discovery of this small molecule (Shigemori et al., 1991). Both failed to show a response to the drug in terms of NGF secretion, despite their reported sensitivity to the growth inhibitory effects of fellutamide B. Likewise, the preneuronal cell line PC-12 (rat pheochromocytoma) which can differentiate into a neuronal phenotype in the presence of NGF (Obin et al., 1999) also failed to show detectable NGF secretion in the presence of fellutamide B either by ELISA (Table 1) or by neurite outgrowth (unpublished observation). Fellutamide B causes cell cycle arrest (measured by [3H]-thymidine incorporation into cellular DNA) in all the cell lines on which it was tested (Table 1). In most cell lines, this growth arrest resulted from the cytoxicity of fellutamide B (measured by MTS conversion by active mitochondria in Table 1), although in a few instances – L-M cells, and to lesser extent C6-2B and A172 cells – the IC50 for growth arrest was below that observed for cytotoxicity. Thus, while the growth inhibitory/cytotoxic activity of fellutamide B appears to affect all cells, its activity to elicit NGF upregulation is restricted to a subset of cell types.
Table 1.
Biological activities of fellutamide B at various cell lines. Fellutamide B treatment was for 24 hr. and values reported are the mean ± standard error of at least three independent experimental determinations.
| Cell Line | Tissue Type | NGF secretion: vehicle-treated (0.1% DMSO) | NGF secretion: 10 µM fellutamide B-treated | Growth Arrest ([3H]-thymidine) IC50 | Cytotoxicity (MTS conversion) IC50 |
|---|---|---|---|---|---|
| S180 | sarcoma | 1.1 ± 0.4 ng/ml | 4.5 ± 1.5 ng/ml | 3.39 ± 0.59 µM | 4.12 ± 0.34 µM |
| NIH-3T3 | fibroblast | 0.3 ± 0.2 ng/ml | 4.7 ± 2.0 ng/ml | 3.40 ± 0.79 µM | 4.62 ± 0.44 µM |
| L-M | fibroblast | 3.0 ± 1.6 ng/ml | 50.3 ± 14.9 ng/ml | 397 ± 44.9 nM | 2.24 ± 0.28 µM |
| A172 | glioblastoma | 19.4 ± 4.7 pg/ml | 95.9 ± 35.8 pg/ml | 347 ± 59.1 nM | 852 ± 77.5 nM |
| C6-2B | glioma | 9.4 ± 4.4 pg/ml | 61.7 ± 29.1 pg/ml | 473 ± 143 nM | 1.52 ± 0.25 µM |
| KB | epidermoid carcinoma | none detected | none detected | 257 ± 46.1 nM | 342 + 82.8 nM |
| P-388 | leukemia | none detected | none detected | 4.26 ± 0.85 µM | 7.80 ± 1.9 µM |
| PC12 | pheochromocytoma | none detected | none detected | 403 ± 19.9 nM | 565 ± 106 nM |
Since L-M cells displayed the most robust NGF response, this cell line was selected for further analysis of fellutamide B activities: as shown in Figure 3a, the potency with which fellutamide B induces NGF in L-M cells is similar to its cytotoxicity in MTS conversion assay (Figure 3b). The bond formed between proteasome catalytic subunits and peptide aldehyde inhibitors like fellutamide B is reversible. Thus, it was hypothesized that a brief administration of fellutamide B for a few hours (a “pulse”) followed by a drug-free “recovery” interval -- rather than a continuous 24 hr. fellutamide B treatment -- might result in significant NGF induction with reduced cytotoxicity compared to continuous drug treatment. It was found that 7 hr. pulse of 10 µM fellutamide B was sufficient to elicit 90% of the maximal NGF upregulation that had been induced by 24 hr of continuous treatment (Figure 3c). However, the reduction in cell viability (Figure 3d) associated with the 7 hr pulse was only 35.0%, which was markedly reduced compared to the 60.0% reduction seen with the continuous 24 hr treatment. Thus, although an exclusively neurotrophic and non-toxic concentration of fellutamide B was not apparent, the two activities of fellutamide B can yet be largely dissociated from each other by the proper duration of treatment.
Figure 3. Treatment of L-M cells with fellutamide B or other proteasome inhibitors induces secretion of NGF.
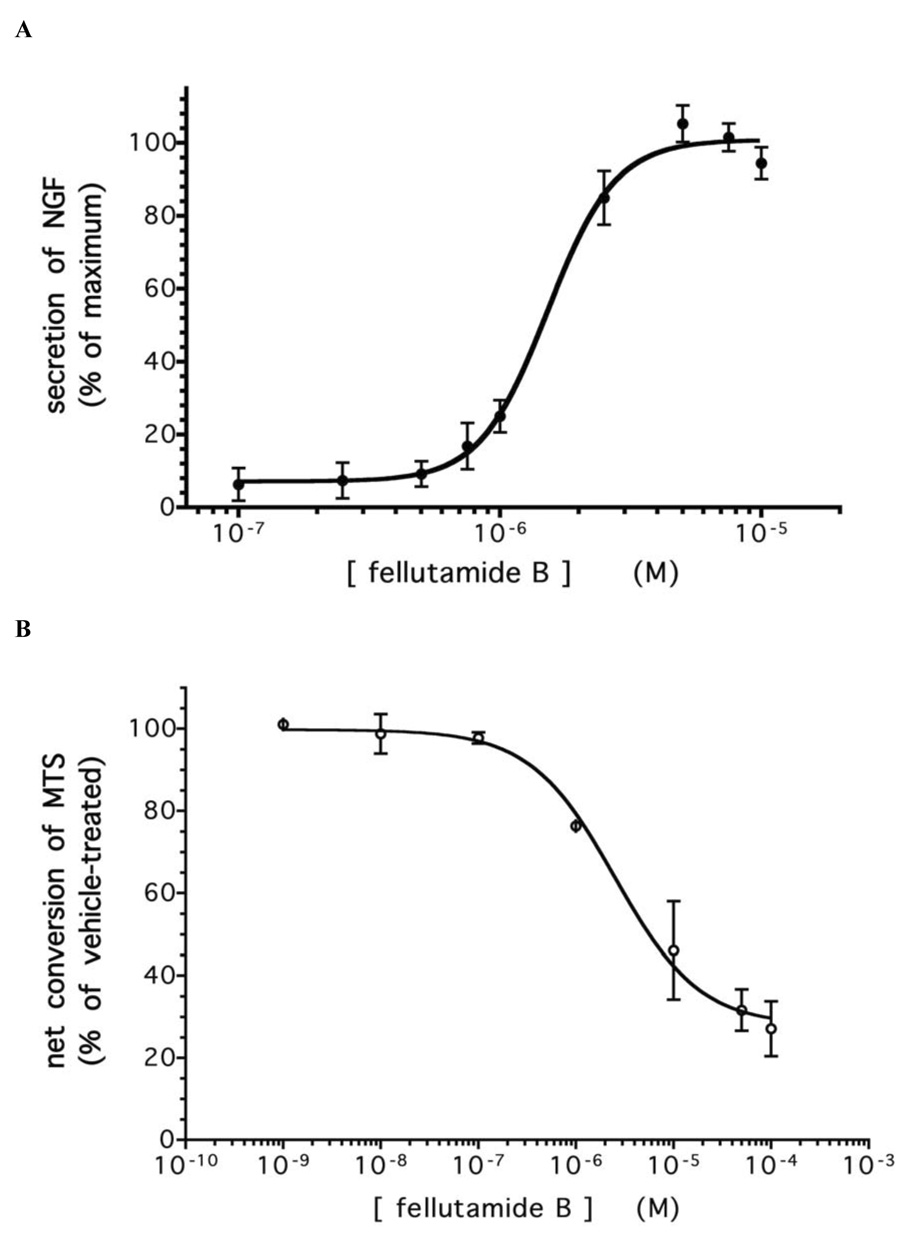
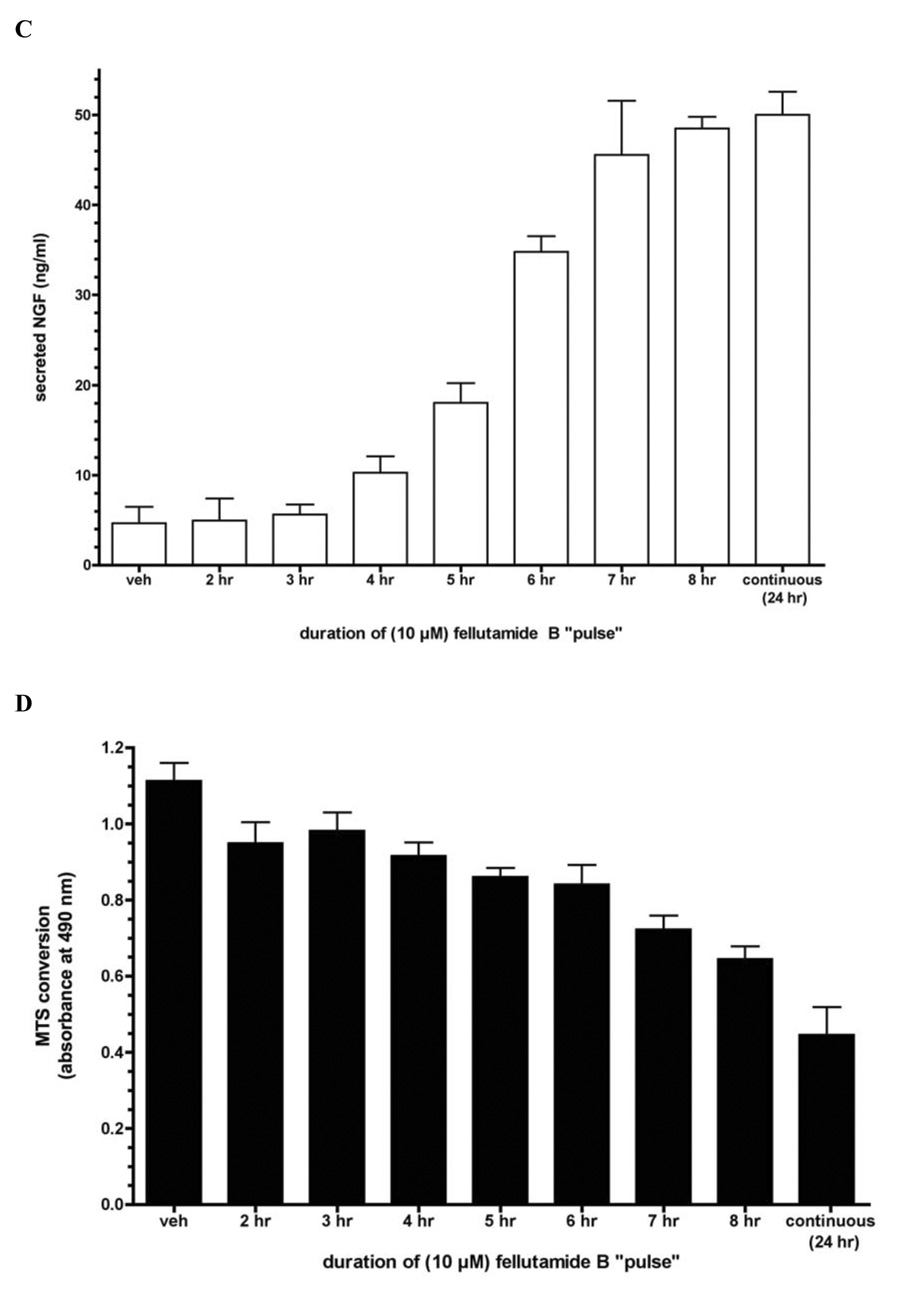


A. Fellutamide B treatment for 24 hr. induces dose-dependent secretion of NGF from L-M cells. B Cytotoxicity of 24 hr. fellutamide B treatment against L-M cells. C. Effectiveness of short (“pulse”) fellutamide B treatments to induce NGF upregulation in L-M cells. D. Cytotoxicity of short (“pulse”) fellutamide B treatments against L-M cells. E. Epoxomicin induces secretion of NGF from L-M cells. F. MG132 induces secretion of NGF from L-M cells. Data presented are the means ± standard deviation of three independent experiments. G. Conditioned medium from L-M cells treated with proteasome inhibitors causes differentiation of preneuronal PC12 cells. Conditioned medium from vehicle-treated L-M cells (top panel); from 10 µM fellutamide B-treated L-M cells (center panel); and from 250 nM epoxomicin-treated L-M cells (bottom panel). Representative images presented.
Having now confirmed the NGF-inducing activity of fellutamide B, and having also shown it to be a proteasome inhibitor, we tested whether this NGF-inducing activity could be generalized to other proteasome inhibitors. Similar to fellutamide B, the established proteasome inhibitors epoxomicin and MG132 triggered NGF upregulation in the L-M cells with EC50 values of 127 ± 13.4 nM and 11.8 ± 3.7 µM, respectively (Figures 3e and 3f). This confirms that proteasome inhibition is the key activity of fellutamide B, since epoxomicin is a highly specific inhibitor of the proteasome that does not inhibit other proteases. While we observed that fellutamide B and the other proteasome inhibitors less potently induced upregulation of NGF than they inhibited the purified proteasomes, this is likely due to the inherent differences between the two assays. Intact cells were required in the NGF secretion assay (as well as the anti-ubiquitin westerns in Figure 1d), and thus factors like membrane permeation, drug-efflux pumps (e.g. P-glycoprotein) and de-ubiquitinases that often reduce the apparent potency of these drugs would have attenuating effect; such factors are absent from our measurements on the purified proteasome activity, which cannot be accurately, specifically made in intact cells due to the interfering presence of free proteases. Nevertheless, the rank order potency with which these compounds induce NGF secretion corresponds to their rank order potency for inhibiting the proteasomal chymotryptic-like activity. The biological activity of the secreted NGF was confirmed by incubating undifferentiated PC12 cells in medium that had been conditioned by fellutamide B- or epoxomicin-treated L-M cells. PC12 neurite outgrowth was readily observable after only 24 hrs. incubation (not shown) and pronounced by 48 hrs. incubation in conditioned medium (Figure 3g).
Given the cytotoxicity accompanying fellutamide B treatment, the question remained whether NGF induction was a consequence of proteasome inhibition specifically or rather a response to general cytotoxic insult. To answer this, L-M cells were separately exposed (Figure 4a) to three different toxic treatments: 1) a combination of veratridine and ouabain, to collapse the membrane potential; 2) a combination of cytochalasin D and myoseverin, to destroy the cytoskeleton; and 3) epoxomicin, a structurally dissimilar proteasome inhibitor. In the case of fellutamide B and epoxomicin, these agent(s) induced the formation of long cell processes which in the past had been misinterpreted as neurites (Fenteany et al., 1994) when observed on neuronal cell lines. All treatments were comparably cytotoxic as measured by MTS conversion assay (Figure 4b), which western blotting for cleavage of the caspase 3 substrate PARP (Figure 4c) showed was due to apoptotic cell death. However, only fellutamide B and epoxomicin triggered upregulation of NGF (Figure 4d). Thus, the neurotrophic effects of fellutamide B appear to be a specific consequence of proteasome inhibition rather than a cellular response to general cytotoxicity.
Figure 4. Secretion of NGF is not a response to general cytotoxicity.



A. Morphological changes to L-M cells in response to 24 hr. treatment with depicted toxins (representative images presented) B. MTS conversion assay of L-M cells treated overnight with indicated toxins. C. Fellutamide B and other toxins trigger apoptotic cleavage of poly(ADP-ribose) polymerase (treatment for 24 hr.). D. ELISA measurements of secreted NGF from L-M cells treated overnight with indicated toxins. Data presented are the means ± standard deviation of three independent experiments.
Upregulation of NGF occurs by induction of new gene transcription from both NGF promoters
To provide further mechanistic insight into proteasome inhibitor-induced NGF upregulation, we next examined the effect of fellutamide B on NGF gene transcription. Specifically, the levels of NGF mRNA from fellutamide B- versus vehicle-treated cells were measured using RT-PCR. These results (Figure 5a) showed a robust upregulation of NGF mRNA in response to the proteasome inhibitor, while measurements of the housekeeping transcript GAPDH showed no drug-associated changes. There are, in fact, two known promoters for the NGF gene (Racke et al., 1996): one upstream of the first exon, and another that lies in between exons 2 and 3, with the entire protein coding region located in exon 4. The mRNAs produced from the two NGF promoters can be distinguished by their differentially retained exons. Our RT-PCR results show that NGF mRNA transcripts from both known promoters are strongly upregulated following fellutamide B treatment (Figure 5a); note that two mRNA transcripts of slightly different sizes are detected from the upstream promoter, as previously seen (Racke et al., 1996). The magnitude of this increase in NGF mRNA following proteasome inhibitor treatment may be sufficient to account for the increases in NGF at the protein level. To confirm that the upregulation of NGF was due to NGF mRNA transcription, a time course of fellutamide-triggered NGF induction in the absence/presence of the RNA polymerase II inhibitor, α-amanitin, was performed. Following 12 hr. of continuous fellutamide B treatment, the levels of secreted NGF began to rise (Figure 5b); this was completely blocked at all times by the presence of α-amanitin. This result is consistent with upregulation due to de novo NGF mRNA synthesis due upon fellutamide B administration, and rules out any independent role for post-translational upregulation of NGF secretion. A parallel MTS assay showed conclusively that the inhibition of NGF upregulation by α-amanitin cannot be attributed to additive cytotoxicity in combination with fellutamide B, as there was none (Figure 5c). Furthermore, measurements of NGF mRNA stability in the absence or presence of fellutamide B (Figure 5d) showed no differences in rate of decay, demonstrating that upregulation of NGF mRNA in response to the small molecule was due only to enhanced transcription and not diminished transcript degradation.
Figure 5. Proteasome inhibition causes upregulation of NGF gene transcription.


A. RT-PCR for NGF and GAPDH transcripts from extracted mRNA from 24 hr. fellutamide B-treated and untreated cells: total NGF mRNA (top left), GAPDH (bottom left), NGF mRNA transcribed from upstream promoter (top, right), and NGF mRNA transcribed from downstream promoter (bottom, right). Representative results shown. B. Blockade of RNA polymerase II with α-amanitin (20 µg/ml) abolishes upregulation of NGF secretion by fellutamide B (10 µM). White bars correspond to control cells, black bars to fellutamide B-treated cells, and grey bars to fellutamide B + α-amanitin-treated cells. C. α-Amanitin and fellutamide B co-treatment does not result in additive cytotoxicity to L-M cells. Colored bars represent the same drug treatment as in B. For B and C, data presented are the means ± standard deviation of three independent experiments. D. Upregulation of NGF mRNA levels by fellutamide B does not involve enhanced stabilization of NGF mRNA transcripts. RT-PCR was performed on mRNA isolated from L-M cells treated with α–amanitin (20 µg/ml) and maintained with or without 10 µM fellutamide B for the times indicated. A representative time-dependent decay of NGF mRNA from vehicle-treated (top panel) and fellutamide B-treated (bottom panel) cells is shown.
In order to focus in on the region(s) of the NGF promoter mediating the transcriptional activation response to proteasome inhibitors, we subcloned the entire 5 kb mouse NGF promoter (D'Mello and Heinrich, 1991) including the first 120 bp downstream of the transcription start site (defined as position +1) into a promoter-less firefly luciferase plasmid for gene reporter assays. Upstream regions of the NGF promoter were progressively removed to create plasmids (Figure 6a) which were stably transfected into cells, along with SV40 early promoter-driven renilla luciferase co-reporter, and tested for the ability of fellutamide B to induce firefly luciferase activity (Figure 6b). To ensure that any effects on reporter expression/regulation arising from differing genomic integration points would be balanced out between the different reporter transfectants, pools of stably-transfected clones for each reporter were used. The full 5 kb NGF promoter (−5000 bp) was responsive to fellutamide B, showing a nearly six-fold induction of luciferase activity. While truncation of the promoter down to 1.8 kb (−1800 bp) increased overall luciferase transcription (basal and drug-induced), the level of induction by fellutamide B remained approximately the same. Further truncation of the promoter down to 750 bp (−750 bp), and then 250 bp (−250 bp) continued the trend of increasing overall luciferase transcription, indicating the removal of postulated gene suppressive elements from the promoter (D'Mello and Heinrich, 1991). Yet, these reporters were still responsive to fellutamide B and showed increased luciferase activity in its presence. Given the importance of the AP-1 site at position +35 in mediating the stimulatory effects of phorbol esters (Omae et al., 1994) and dihydroxyvitamin D3 (Veenstra et al., 1998) on NGF synthesis, and the fact that fellutamide B treatment causes the stabilization and activation of c-Jun (unpublished observation), we tested whether this AP-1 site is also crucial for the effects of proteasome inhibitors. Reporter vectors were constructed lacking the AP-1 site (−1800 w/o AP-1; −750 w/o AP-1; and −250 w/o AP-1), but assays on cells transfected with these constructs continued to show increased luciferase activity in response to fellutamide B, albeit removal of the AP-1 site had a diminishing effect on overall luciferase transcription. Truncation of the promoter down to 150 bp retained fellutamide B responsiveness, indicating that the cis-acting element or elements lie close to the transcription start site. Epoxomicin, MG 132 and clasto-lactacystin β-lactone all induced luciferase (Figure 6c) from this minimal reporter plasmid, verifying that this region of the NGF promoter was responsive to proteasome inhibitors other than fellutamide B.
Figure 6. A cis-acting element within the NGF promoter is induced by proteasome inhibitors.


A. Schematic of NGF promoter-driven luciferase reporters. White regions represent the NGF promoter and black regions represents firefly luciferase gene (not to scale). B. Fellutamide activates a cis-acting element adjacent to the transcription start site. Data presented are NGF promoter-driven firefly luciferase reporter activity normalized to SV40 early promoter-driven renilla luciferase co-reporter. Results are the mean ± standard error of 3 to 5 independent experiments. C. Proteasome inhibitors other than fellutamide B induce via the same cis-acting element in the NGF promoter. Cells were stably-transfected with the luciferase reporter driven by the 150 bp upstream of the transcription start site in the NGF promoter (a.k.a. “-150 bp w/o AP1”). Results are the mean ± standard error of 3 to 5 independent experiments.
DISCUSSION
Our finding that proteasome inhibition leads to the production and secretion of NGF reveals another potential therapeutic utility of this class of small molecules. Proteasome inhibitors have already been identified as candidate anti-inflammatory (Meng et al., 1999b) and anti-tumor drugs (Meng et al., 1999a). They have also been implicated in stimulating bone formation (Garrett et al., 2003), the treatment of stroke (Phillips et al., 2000), and as antiparasitic agents (Lindenthal et al., 2005). While proteasome inhibition can lead to cytotoxicity, therapeutic windows have been established that nonetheless permit their use clinically. Indeed, while a continuous 24 hr treatment of the cells with fellutamide B resulted in maximum NGF upregulation and toxicity, “pulse” treatment with fellutamide B for only 7 hours followed by a 17 hr. recovery interval resulted in a nearly equivalent induction of NGF with markedly reduced toxicity. The separation of these two activities of fellutamide B – and, by extension, other reversible proteasome inhibitors ‒ is an important step in their development as potential neurotrophic therapeutics. This is less of a concern with fellutamide B than it will be with other proteasome inhibitors: epoxomicin was the most potent inducer of NGF, but was also the most potently cytotoxic. This greater toxicity is almost certainly attributable to the fact that epoxomicin, unlike the fellutamide B and MG132, irreversibly inhibits the proteasome, which placed an upper limit on the epoxomicin concentrations that could be used on whole cells (e.g. Figure 1d and Figure 3e) and still produce the biological effect without killing them first. This irreversible inhibition by epoxomicin ultimately makes it unsuitable for the same “pulse” treatments that reduced the toxicity of fellutamide B.
Application of proteasome inhibitors such as MG132 to transected axons significantly delays the onset of Wallerian degeneration (Zhai et al., 2003) both in vitro and in vivo. Given that persistent activation of erk1/2 (MacInnis and Campenot, 2005), a known downstream effector of NGF receptors, was also observed in these studies was, it is possible that the degeneration was delayed by proteasome inhibitor-triggered upregulation of NGF. The ability of some proteasome inhibitors to spur “neurite outgrowth” from preneuronal cells has been reported many times (Fenteany et al., 1994; Inoue et al., 2004); however, this effect is not specific to preneuronal cells, and has been shown to also occur in endothelial cells (Meng et al., 1999a) and, in the present study, in L-M fibroblasts (Figure 4a). Mitchison and colleagues have reported that proteasome inhibitors arrest dividing cells in cytokinesis (Straight et al., 2003), which may be the basis for the spindle-like morphology seen in treated cells. Thus, it is perhaps more accurate to describe this proteasome inhibitor effect as a general induction of a bipolar cellular elongation; which in preneuronal cells was misinterpreted as “neurites”. However, NGF induction by proteasome inhibition is a process that should elicit neuronal differentiation, maintenance and neuroprotection given that it upregulates the natural neurotrophin. Further evidence for the independence of proteasome inhibitor-triggered process extension from NGF secretion is that fellutamide B failed to trigger the latter in PC12 cells (data not shown), a system in which neurite extension following proteasome inhibition has been studied extensively.
The structural data from the co-crystallization of fellutamide B with the 20S proteasome reveals some interesting and unexpected insights into how inhibition is achieved. It illustrates the possibility of two different hemiacetal adduct enantiomers (R,S) formed by peptide aldehyde inhibitors: the customary adduct with the planar aldehyde group oxygen atom pointing into the oxyanion hole formed by Gly47-N; or, as now seen with fellutamide B, an alternative orientation of the aldehyde group oxygen towards Thr1-NH. Although hydrogen bonding between the Thr1-N terminus and the active group of a small molecule inhibitor has been observed for other classes of inhibitors (e.g. β-lactones (Borissenko and Groll, 2007)), it is nonetheless an unexpected finding for a peptide aldehyde proteasomal inhibitor that either orientiation can and will stabilize the hemiacetal oxygen atom and thereby block catalytic activity (Borissenko and Groll, 2007; Groll et al., 1997; Löwe et al., 1995). In addition to the binding of fellutamide B’s P1 and P3 side chains to the cognate S1 and S3 specificity pockets, the structural basis for its preferential blockade of the chymotryptic-like active site may also reside in the interaction of its distinctive aliphatic tail with several residues in an adjacent hydrophobic groove. The aliphatic tail adopts dissimilar conformations when bound to the tryptic-like and caspase-like active sites, such that the stabilization of the aliphatic tail to the hydrophobic groove is peculiar to the chymotryptic-like active site. This may contribute significantly to the tighter binding of fellutamide B to that subunit, translating into the observed 200- to 400-fold greater potency to inhibit this activity (Figure 1b).
An earlier study (Yamaguchi et al., 1993) had shown decreasing NGF secretion triggered by a maximum dose of fellutamide in the presence of ever-increasing concentrations of actinomycin D. This raised two possibilities: either the increasing, combined cytotoxicity from blockade of both the proteasome and transcription killed the cells before NGF could be produced, or de novo NGF mRNA transcription is a necessary element in the mechanism of fellutamide. We have, in fact, observed that fellutamide B and other proteasome inhibitors exert a biphasic effect on NGF secretion: increasing it until a critical concentration of proteasome inhibitor is reached, after which further increases become overwhelmingly toxic and NGF secretion diminishes. Thus, a more rigorous examination of the involvement of mRNA upregulation in the effect of fellutamide B seemed particularly compelling. The data here show conclusively that increased transcription of NGF mRNA is, indeed, necessary – in fact, the increase in NGF mRNA following fellutamide B treatment was so dramatic that it recommended upregulation occurs exclusively at the mRNA level. This increase in NGF mRNA involves activation of both known promoter regions for the NGF gene, consistent with an earlier report suggesting coordinate regulation of these two promoters (Racke et al., 1996). Since the ultimate NGF protein products from these two promoters are believed to be biologically equivalent, the combined effect of proteasome inhibition would be that much greater.
While our own RT-PCR results clearly showed that NGF mRNA was being upregulated by fellutamide B, it was possible that either degradation of the transcripts had been attenuated or their synthesis had been stimulated, or some combination of both. The blockade of the neurotrophic response to fellutamide B by α-amanitin suggested transcriptional activation of the NGF gene and ruled out the possibility that increased NGF secretion could be largely post-translational (i.e. NGF protein trafficking). Still, it did not eliminate the possibility that stabilization of NGF mRNA contributed to the transcript upregulation. Only after showing that the decay of NGF mRNA remains the same in the presence or absence of fellutamide B could we unambiguously conclude that transcriptional activation of the NGF gene is the sole mechanism for increased production of the protein. Furthermore, we were able to narrow the region of the NGF promoter crucial for transcriptional activation down to within 150 bp of the transcription start site and rule out participation of an important AP-1 site at position +35.
Changes in levels of other proteins besides NGF are to be expected following treatment with proteasome inhibitors, which is why these small molecules have therapeutic value for treating many disease states. However, the broad effects of these small molecules can also lead to potential limitations on their usefulness. For example, some uncertainty exists regarding the stabilizing effect of proteasome inhibition on cytoplasmic levels of the non-infectious form of the prion protein, PrPC. While transmission of prion disease states depends on the presence of the misfolded “scrapie” form of the protein (PrPSc), Lindquist and colleagues reported that accumulation of PrPC following proteasome inhibition can itself result in neuronal toxicity (Ma and Lindquist, 2001). Although subsequent reports (Fioriti et al., 2005; Kristiansen et al., 2005; Roucou et al., 2003) strongly dispute the neurotoxic effect of elevated cytoplasmic PrPC, the effects of proteasome inhibitors on protein levels other than NGF need to be carefully considered as their potential for neuronal therapeutics is evaluated. Separating the toxic effect of fellutamide B and other proteasome inhibitors from their NGF-inducing properties is a crucial barrier towards their ultimate development for any clinical use, neurotrophic or otherwise. While it was demonstrated that toxicity is not a precondition for upregulating NGF from the cell types tested, toxicity is a side effect of proteasome inhibitors. There are, however, reasons to believe that these two activities of fellutamide B are independent. For instance, not every cell tested herein responded to fellutamide B by upregulating NGF; yet all the cells tested were sensitive to the cytotoxic effect of the proteasome inhibitor. In addition, the aforementioned pulse-recovery strategy of administering fellutamide B produced near maximum NGF upregulation with attenuated toxicity. Moreover, the dose-response relationship of fellutamide B cytotoxicity clearly differs from that of NGF induction in that the latter reaches maximum efficacy over a more narrow concentration range: as shown in Figure 3b, the Hill slope for fellutamide B cytotoxicity fits to unity, while that for its NGF induction (as well as MG132 and epoxomicin) is 3.5 (as shown in Figures 3a, e and f). While the molecular basis for this pharmacodynamic difference is not known, it suggests the possibilities of positive cooperativity or other positive feedback mechanism in the upregulation of NGF by fellutamide B or the activation of multiple cis-elements over a narrow concentration range of fellutamide B (non-first order induction); this is not seen with cytotoxicity. Since the blockade of the proteasome affects the levels of multiple proteins within the cell, it is reasonable to hypothesize that the protein(s) that cause upregulation of NGF production will be different from those that mediate the toxic effect. Thus, maximum NGF induction can be achieved at proteasome inhibitor conditions that do not elicit maximum toxicity.
It is encouraging that fellutamide B exerts its effects on both glial-type and fibroblast-type cells. In terms of therapeutic potential, a broader tissue effect would enable proteasomal inhibition to treat both peripheral nerve injury, where fibroblasts would be the predominant cell type to secrete the trophic factors, as well as neurodegeneration in the brain, where glial type cells are more plentiful. Moreover, as many neurotrophins appear to be coordinately regulated in the CNS (Maisonpierre et al., 1990; Takeda et al., 1993), the possibility that proteasome inhibition might have a similar trophic effect on other neurotrophins (e.g. NT-3, BDNF) is an interesting possibility that remains to be investigated.
SIGNIFICANCE
There is great interest in the fields of neuroscience and chemical biology to identify compounds that either trigger neuronal differentiation and survival or induce neurotrophin expression. Such compounds would have great therapeutic potential for the treatment of neuronal injury or neurodegenerative diseases. While reports that identify such compounds from library screens or natural sources are increasing in frequency, none of these emerging studies have identified a protein receptor and/or mechanism of action to which the neurotrophic activity can be ascribed. We report here that fellutamide B, one of the earliest compounds identified to have neurotrophin-inducing activity, does so by binding to and inhibiting the 20S proteasome. While the manner in which fellutamide B binds to the proteasome is distinct from other peptide aldehyde inhibitors, its ability to upregulate NGF is shared not only by other peptide aldehydes, but other mechanistically different classes of proteasome inhibitors as well. Proteasome inhibitors as well as NGF administration are known to attenuate Wallerian axonal degeneration and our results now suggest a direct connection between these two neuroprotective approaches.
The proteasome has previously been targeted in the treatment of cancer, inflammation and stroke – herein, we have identified another potential medical application for proteasome inhibitors, some of which are already in clinical trials. With the demonstrated efficacy of neurotrophins to alleviate symptoms of neurodegenerative diseases, fellutamide B and other proteasome inhibitors deserve further attention as potential neuronal therapeutics.
EXPERIMENTAL PROCEDURES
Reagents
Fellutamide B (Schneekloth et al., 2006) and epoxomicin (Sin et al., 1999) were synthesized as previously described. MG 132, myoseverin, cytochalasin D, clasto-lactacystin β-lactone and α-amanitin were purchased from Calbiochem (La Jolla, CA). Veratridine and ouabain were obtained from Sigma (St. Louis, MO). Fluorogenic substrate peptides for proteasome activity assay were from Bachem Bioscience (King of Prussia, PA). Purified human 20S proteasome were purchased from Boston Biochemical (Cambridge, MA)
Cell culture
C6-2B cells were a generous gift from Satya Kunapuli (Temple University, Philadelphia, PA), and PC12 cells were a gift from Randy Pittman (University of Pennsylvania, Philadelphia, PA); all other cell lines were purchased from ATCC (Manassas, VA). Undifferentiated PC12 cells were grown in RPMI 1640 medium supplemented with 10% heat-inactivated horse serum, 5% heat-inactivated fetal bovine serum. L-M mouse fibroblasts were cultured in Medium 199 supplemented with 0.5% peptone. NIH-3T3, C6-2B and A172 cells were grown in high glucose Dulbecco’s Modified Eagle Medium supplemented with 10% heat-inactivated fetal bovine serum and S-180 cells were cultivated in minimal essential medium supplemented with Earle’s salts, 2 mM glutamine, 1 mM sodium pyruvate and 0.1 mM non-essential amino acids. All culture media was supplemented with 100 units/ml penicillin G and 100 µg/ml streptomycin sulfate.
Cytotoxicity assay
Following drug treatment of cells as indicated, culture medium was supplemented with 330 µg/ml MTS (Promega Corp., Madison, WI) and 25 µM phenazine methosulfate and incubated at 37°C. Mitochondrial reduction of MTS to the formazan derivative was monitored by measuring the medium’s absorbance at 490 nm.
Proliferation assay
Performed as previously described (Yeh et al., 2000). [3H]-Thymidine was purchased from Perkin-Elmer Life Sciences (Boston, MA).
Neurite outgrowth assay
L-M cells were treated with either 10 µM fellutamide B, 250 nM epoxomicin, or vehicle control (0.1% DMSO) for 24 hr. Afterwards conditioned medium was collected and dialyzed (MWCO = 10 kDa) against RPMI 1640 for 24 hrs. at 4°C. The dialyzed, conditioned medium was then supplemented with 2% heat-inactivated horse serum and 1% heat-inactivated fetal bovine serum and applied to undifferentiated PC12 cells growing on collagen-covered 60 mm dishes. The development of neurites was monitored over the following 48 hrs.
NGF ELISA
ELISA for NGF was performed in accordance with the kit manufacturer’s recommended instructions (Promega Corp., Madison, WI). Data were analyzed using PRISM software (GraphPad Software, San Diego, CA)
Western Blotting
Performed as previously described (Meng et al., 1999a). Protein samples were resolved by 8% SDS-PAGE, transferred to nitrocellulose and probed with antibodies to ubiquitin (Cell Signaling Tech., Danvers, MA), α-tubulin (Sigma, St. Louis, MO) or PARP (Zymed Labs, San Francisco, CA).
Proteasome activity assay
Performed according to previously described detailed method (Kim et al., 2005).
RT-PCR
mRNA was extracted from fellutamide B-treated and vehicle-treated L-M cells using Trizol reagent. First strand synthesis of cDNA was performed using SuperScript III first strand synthesis kit (Invitrogen, Carlsbad, CA). PCR measurements of NGF and GAPDH were performed using the following primers:
total NGF forward primer: 5’-GCAGTGAGGTGCATAGCGTA-3’
upstream promoter-derived NGF forward primer: 5’-AGAGAGCGCCTGGAGCCG-3’
downstream promoter-driven NGF forward primer: 5’-CTTCCTGGGCTCTAATGATGC-3’
total NGF reverse primer: 5’-CACTGAGAACTCCCCCATGT-3’
GAPDH forward primer: 5’-AACTTTGGCATTGTGGAAGG-3’
GAPDH reverse primer: 5’-ACACATTGGGGGTAGGAACA-3’
PCRs were performed for 30 cycles using decreasing titrations of template cDNA to verify changes (or lack thereof) in target abundance.
Co-crystallization of fellutamide B and 20S proteasome
Crystals of 20S proteasome from S. cerevisiae were grown in hanging drops at 24°C as described previously (Groll and Huber, 2005) and incubated for 60 min with fellutamide B (10 mM in DMSO). The protein concentration used for crystallization was 40 mg/ml in 10 mM Tris.HCl, pH 7.5, and 1 mM EDTA. Drops contained 3 µl of protein and 2µl of reservoir solution (30 mM magnesium acetate, 100 mM morpholino-ethane-sulphonic acid (MES) pH 7.2, and 10% 2-methyl-2,4-pentanediol (MPD)). The space group of proteasomal complex crystals belongs to P21 with cell dimensions of a = 134.3 Å, b = 301.6 Å, c = 143.5 Å and β = 112.7°. Data to 2.6 Å were collected using synchrotron radiation with λ = 1.05 Å at the BW6-beamline at DESY, Hamburg, Germany. Crystals were soaked in cryoprotecting buffer (30% MPD, 20 mM magnesium acetate, 100 mM MES, pH 6.9) and frozen in a stream of liquid nitrogen gas at 90 K (Oxford Cryo Systems, Oxford, UK). X-ray intensities were evaluated using the DENZO program package and data reduction was performed with SCALEPACK (Otwinowski et al., 2003; Otwinowski and Minor, 1997). Anisotropy of diffraction was corrected by an overall anisotropic temperature factor, comparing observed and calculated structure amplitudes using the program CNS (Brünger et al., 1998). A total of 796875 reflections yielded 331875 unique reflections (98.7% completeness). The corresponding Rmerge was 6.6% at 2.6 Å resolution (40.2% for the last resolution shell). Electron density was improved by averaging and back-transforming reflections 10 times over the two-fold non-crystallographic symmetry axis using the program package MAIN (Turk, 1992). Conventional crystallographic rigid body, positional and temperature factor refinements were carried out with CNS using the yeast 20S proteasome structure as the starting model (Groll et al., 1997). For model building the program MAIN was used. The structure was refined to a crystallographic R-factor of 24.0% (free R-factor 26.5%(Brünger, 1993)) with rms-deviations from target values of 0.007 Å for bonds and 1.37° for angles(Brünger, 1993). Modeling experiments were performed using the coordinates of yeast 20S proteasome (Groll et al., 1997) with the program MAIN.
Luciferase reporter promoter activity assay
Using primer directed PCR, truncated versions of the previously cloned NGF promoter were created and inserted into the promoterless luciferase expression vector pGL4 (Promega Corp.). Reporter constructs were then stably transfected into NIH-3T3 cells using LipofecatAMINE 2000; pools of stably-transfected clones were used for the analysis of each reporter construct. The stably-transfected cells were tested for their ability to upregulate luciferase activity in response to proteasome inhibitors using the Dual Luciferase Reporter Assay System (Promega Corp.). An SV40 early promoter-driven renilla luciferase construct (Promega Corp.) was used as a co-reporter to normalize results.
ACKNOWLEDGMENTS
The authors acknowledge the NIH (GM062120) for funding.
Footnotes
COMPETING INTERESTS STATEMENT
C.M.C. is a co-founder of Proteolix, Inc., which currently is exploring the use of proteasome inhibitors in oncology.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Borissenko L, Groll M. 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chem. Rev. 2007;107:687–717. doi: 10.1021/cr0502504. [DOI] [PubMed] [Google Scholar]
- Brünger A, Adams P, Clore G, DeLano W, Gros P, Grosse-Kunstleve R, Jiang J, Kuszewski J, Nilges M, Pannu N, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;1:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Brünger AT. Assessment of phase accuracy by cross validation: the free R value. Methods and applications. Acta Crystallogr. D Biol. Crystallogr. 1993;49:24–36. doi: 10.1107/S0907444992007352. [DOI] [PubMed] [Google Scholar]
- Castellanos-Ortega MR, Cruz-Aguado R, Martinez-Martí L. Nerve growth factor: possibilities and limitations of its clinical application. Rev. Neurol. 1999;29:439–447. [PubMed] [Google Scholar]
- Chaturvedi RK, Shukla S, Seth K, Agrawal AK. Nerve growth factor increases survival of dopaminergic graft, rescue of nigral dopaminergic neurons and restores functional deficits in rat model of Parkison's disease. Neurosci. Lett. 2006;398:44–49. doi: 10.1016/j.neulet.2005.12.042. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Schneider B, Riese U, Schubert B, Li Z, Hamburger M. (+)-N-Deoxymilitarinone A, a neuritogenic pyridone alkaloid from the insect pathogenic fungus, Paecilomyces farinosus. J. Nat. Prod. 2006;69:436–438. doi: 10.1021/np050418g. [DOI] [PubMed] [Google Scholar]
- D'Mello SR, Heinrich G. Structural and functional identification of regulatory regions and cis elements surrounding the nerve growth factor gene promoter. Mol. Brain. Res. 1991;11:255–264. doi: 10.1016/0169-328x(91)90034-u. [DOI] [PubMed] [Google Scholar]
- Fenteany G, Standaert RF, Reichard GA, Corey EJ, Schreiber SL. A β-lactone related to lactacystin induces neurite outgrowth in a neuroblastoma cell line and inhibits cell cycle progression in an osteosarcome cell line. Proc. Natl. Acad. Sci. USA. 1994;91:3358–3362. doi: 10.1073/pnas.91.8.3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioriti L, Dossena S, Stewart LR, Stewart RS, Harris DA, Forloni G, Chiesa R. Cytosolic prion protein (PrP) is not toxic in N2A cells and primary neurons expressing pathogenic PrP mutations. J. Biol. Chem. 2005;280:11320–11328. doi: 10.1074/jbc.M412441200. [DOI] [PubMed] [Google Scholar]
- Garrett IR, Chen D, Gutierrez G, Zhao M, Escobedo A, Rossini G, Harris SE, Gallwitz W, Kim KB, Hu S, et al. Selective inhibitors of the osteoblast proteasome stimulate bone formation in vivo and in vitro. J. Clin. Invest. 2003;111:1771–1782. doi: 10.1172/JCI16198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R. Structure of 20S proteasome from yeast at 2.4 Å resolution. Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- Groll M, Huber R. Purification, crystallization and X-ray analysis of the yeast 20S proteasomes. Methods Enzymol. 2005;398:329–336. doi: 10.1016/S0076-6879(05)98027-0. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Ann. Rev. Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Inoue M, Zhai H, Sakazaki H, Furuyama H, Fukuyama Y, Hirama M. TMC-95A, a reversible proteasome inhibitor, induces neurite outgrowth in PC12 cells. Bioorg. Med. Chem. Lett. 2004;14:663–665. doi: 10.1016/j.bmcl.2003.11.043. [DOI] [PubMed] [Google Scholar]
- Kim K-B, Fonseca FN, Crews CM. Development and characterization of proteasome inhibitors. In: Deshaies RJ, editor. Methods in Enzymology v.399. London, U.K.: Elsevier Academic Press; 2005. pp. 585–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansen M, Messenger MJ, Klöhn P-C, Brandner S, Wadsworth JDF, Collinge J, Tabrizi SJ. Disease-related prion protein forms aggresomes in neuronal cells leading to caspase activation and apoptosis. J. Biol. Chem. 2005;280:38851–38861. doi: 10.1074/jbc.M506600200. [DOI] [PubMed] [Google Scholar]
- Lapchak PA. Nerve growth factor pharmacology: application to the treatment of choliergic neurodegeneration in Alzheimer's disease. Exp. Neurol. 1993;124:16–20. doi: 10.1006/exnr.1993.1168. [DOI] [PubMed] [Google Scholar]
- Liao GS, Li XB, Zhang CY, Shu YY, Tang SX. Pharmacological actions of nerve growth factor-transferrin conjugate on the central nervous system. J. Nat. Toxins. 2001;10:291–297. [PubMed] [Google Scholar]
- Lindenthal C, Weich N, Chia YS, Heussler V, Klinkert MQ. The proteasome inhibitor MLN-273 blocks exoerythrocytic and erythrocytic development of Plasmodium parasites. Parasitology. 2005;131:37–44. doi: 10.1017/s003118200500747x. [DOI] [PubMed] [Google Scholar]
- Loidl G, Groll M, Musiol HJ, Huber R, Moroder L. Bivalency as a principle for proteasome inhibition. Proc. Natl. Acad. Sci. USA. 1999;96:5418–5422. doi: 10.1073/pnas.96.10.5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löwe J, Stock D, Jap B, Zwicki P, Baumeister W, Huber R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 Å resolution. Science. 1995;268:533–539. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- Ma J, Lindquist S. Wild-type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. Proc. Natl. Acad. Sci. USA. 2001;98:14955–14960. doi: 10.1073/pnas.011578098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacInnis BL, Campenot RB. Regulation of Wallerian degeneration and nerve growth factor withdrawal-induced pruning of axons of sympathetic neurons by the proteasome and the MEK/Erk pathway. Mol. Cell. Neurosci. 2005;28:430–439. doi: 10.1016/j.mcn.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD. NT-3, BDNF and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- Meng L, Kwok BHB, Sin N, Crews CM. Eponemycin exerts its antitumor effect through the inhibition of proteasome function. Cancer Res. 1999a;59:2798–2801. [PubMed] [Google Scholar]
- Meng L, Mohan R, Kwok BHB, Elofsson M, Sin N, Crews CM. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc. Natl. Acad. Sci. USA. 1999b;96:10403–10408. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung J, Kim KB, Lindsten K, Dantuma NP, Crews CM. Lack of proteasome active site allostery as revealed by subunit-specific inhibitors. Mol. Cell. 2001;7:411–420. doi: 10.1016/s1097-2765(01)00188-5. [DOI] [PubMed] [Google Scholar]
- Obin M, Mesco E, Gong X, Haas AL, Joseph J, Taylor A. Neurite outgrowth in PC12 cells. J. Biol. Chem. 1999;274:11789–11795. doi: 10.1074/jbc.274.17.11789. [DOI] [PubMed] [Google Scholar]
- Omae F, Katsumata T, Sakuma M, Furukawa Y, Furukawa S. Prolonged alkylcatechol-induced expression of c-Jun proto-oncongene followed by elevation of NGF mRNA in cultured astroglial cells. J. Neurosci. Res. 1994;39:290–297. doi: 10.1002/jnr.490390306. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Borek D, Majewski W, Minor W. Multiparametric scaling of diffraction intensities. Acta Crystallogr. A. 2003;59:228–234. doi: 10.1107/s0108767303005488. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Phillips JB, Williams AJ, Adams J, Elliott PJ, Tortella FC. Proteasome inhibitor PS519 reduces infarction and attenuates leukocyte infiltration in a rat model of focal cerebral ischemia. Stroke. 2000;31:1686–1693. doi: 10.1161/01.str.31.7.1686. [DOI] [PubMed] [Google Scholar]
- Racke MM, Mason PJ, Johnson MP, Brankamp RG, Linnik MD. Demonstration of a second pharmacologically active promoter region in the NGF gene that induces transcription at exon 3. Mol. Brain Res. 1996;41:192–199. doi: 10.1016/0169-328x(96)00096-4. [DOI] [PubMed] [Google Scholar]
- Roucou X, Guo Q, Zhang Y, Goodyer CG, LeBlanc A. Cytosolic prion protein is not toxic and protects against Bax-mediated cell death in human primary neurons. J. Biol. Chem. 2003;278:40877–40881. doi: 10.1074/jbc.M306177200. [DOI] [PubMed] [Google Scholar]
- Schneekloth JS, Sanders JL, Hines J, Crews CM. Neurotrophic peptide aldehydes: solid phase synthesis of fellutamide B and a simplified analog. Bioorg. Med. Chem. Lett. 2006;16:3855–3858. doi: 10.1016/j.bmcl.2006.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemori H, Wakuri S, Yazawa K, Nakamura T, Sasaki T, Kobayashi Ji. Fellutamides A and B, cytotoxic peptides from a marine fish-possessing fungus Penicillium fellutanum. Tetrahedron. 1991;47:8529–8534. [Google Scholar]
- Sin N, Kim K-B, Elofsson M, Meng L, Auth H, Kwok BH, Crews CM. Total synthesis of the potent proteasome inhibitor epoxomicin: a useful tool for understanding proteasome biology. Biorg. Med. Chem. Lett. 1999;9:2283–2288. doi: 10.1016/s0960-894x(99)00376-5. [DOI] [PubMed] [Google Scholar]
- Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ. Dissecting temporal and spatial control of cytokinesis with a myosin II inhibitor. Science. 2003;299:1743–1747. doi: 10.1126/science.1081412. [DOI] [PubMed] [Google Scholar]
- Takeda A, Onodera H, Sugimoto A, Kogure K, Obinata M, Shibahara S. Coordinated expression of messenger RNAs for nerve growth factor, brain-derived neurotrophic factor, and neurotrophin-3 in the rat hippocampus following transient forebrain ischemia. Neuroscience. 1993;55:23–31. doi: 10.1016/0306-4522(93)90451-k. [DOI] [PubMed] [Google Scholar]
- Turk D. Thesis in Biochemistry and Biophysics. Munich, Germany: Technische Universität Müenchen; 1992. Improvement of a program for molecular graphics and manipulation of electron densities and its application for protein structure determination. [Google Scholar]
- Tuszynski MH, Thai L, Pay M, Salmon DP, U HS, Bakay R, Patel P, Blesch A, Vahlsing HL, Ho G, et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat. Med. 2005;11:551–555. doi: 10.1038/nm1239. [DOI] [PubMed] [Google Scholar]
- Veenstra TD, Fahnestock M, Kumar R. An AP-1 site in the nerve growth factor promoter is essential for 1,25-dihydroxyvitamin D3-mediated nerve growth factor expression in osteoblasts. Biochemistry. 1998;37:5988–5994. doi: 10.1021/bi972965+. [DOI] [PubMed] [Google Scholar]
- Venero JL, Hefti F, Knusel B. Trophic effect of exogenous nerve growth factor on rat striatal cholinergic neuros: comparison between intraparenchymal and intraventricular administration. Mol. Pharmacol. 1996;49:303–310. [PubMed] [Google Scholar]
- Warashina M, Hoon Min K, Kuwabara T, Huynh A, Gage FH, Schultz PG, Ding S. A synthetic small molecule that induces neuronal differentiation of adult hippocampal neural progenitor cells. Angew. Chem. Int. Ed. 2006;45:591–593. doi: 10.1002/anie.200503089. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K, Tsuji T, Wakuri S, Yazawa K, Kondo K, Shigemori H, Kobayashi Ji. Stimulation of nerve growth factor synthesis and secretion by fellutamide A in vitro. Biosci. Biotech. Biochem. 1993;57:195–199. doi: 10.1271/bbb.57.195. [DOI] [PubMed] [Google Scholar]
- Yeh J-R, Mohan R, Crews CM. The antiangiogenic agent TNP-470 requires p53 and p21CIP/WAF for endothelial growth arrest. Proc. Natl. Acad. Sci. USA. 2000;97:12782–12787. doi: 10.1073/pnas.97.23.12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Q, Wang J, Kim A, Liu Q, Watts R, Hoopfer E, Mitchison T, Luo L, He Z. Involvement of the ubiquitin-proteasome system in the early stages of Wallerian Degeneration. Neuron. 2003;39:217–225. doi: 10.1016/s0896-6273(03)00429-x. [DOI] [PubMed] [Google Scholar]